Alek already mentioned GBrowse and IGV as the visualization tool. You'll want your reference genome to be the one you compare to. Let me just add that you need to convert your BLAST result into a format that the tools can accept. BED format is a good start. You need to have a few columns in this file format (the first three are mandatory).
chrom, start, end, name, score, ...
If you have a BLAST tabular format, these fields are already present in the results.
queryId, subjectId, percIdentity, alnLength, mismatchCount, \
gapOpenCount, queryStart, queryEnd, subjectStart, subjectEnd, eVal, bitScore
To convert to BED format, assuming query is your contigs and subject is the referece:
subjectID, subjectStart - 1, subjectEnd, queryID:queryStart-queryEnd
You might need to swap subjectStart and subjectEnd, depending on which one is larger. Also note in the final BED file, use tab instead of comma.
I always found another tool, MUMMERPLOT to be quite useful - if you can use a different aligner (MUMMER). I always do MUMMERPLOT after assembly.
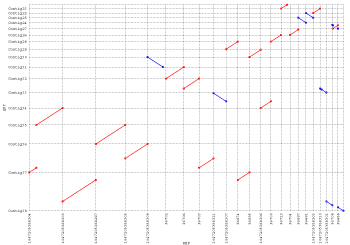




Please fix title "contains" to "contigs".
@Aleksandr, done.