
Hi. I am a biologist in need of a good graphic/visual/fast FastQ editor. Starting from a Biostars thread I implemented few days ago my own SFF/FastQ editor. I hope this is the most complete SFF/FastQ editor available. If you want a specific feature implemented just let me know.
Features
Supported files
- SFF, FastQ, FQ, Fasta (soon)
Filters
- Cut reads with average QV under specified threshold
- Cut reads if they contain N bases (the user can specify how many)
- Cut low complexity reads
- Cut reads that are too short
- Cut reads that are too long
- Cut low quality ends. Automatically detect and cut low quality bases at the end of each read
- Cut poly-A/T tails
{kind=link}
Tools and converters
- Dereplicate sequences (to be released soon!)
- Split multiplexed files (MID/barcode splitter)
- Remove contaminants (search over represented sequences against a contaminant database)
- File splitter: Split huge FastQ/SFF file in chunks of x reads
- File splitter: Cut all sequences in the specified range
- Compact FastQ files
- Convert SFF to FastQ
- Convert SFF to Fasta
- Convert FastQ to Fasta (multiFasta)
- Convert FastQ file to a different encoding (under development)
Graphs and data analysis
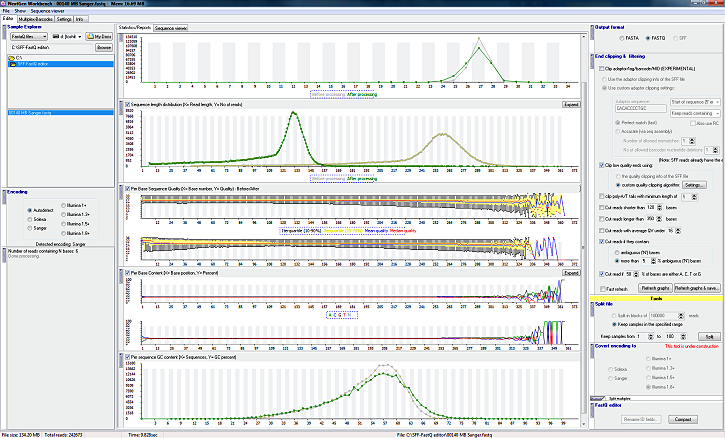
- Sequence viewer - Show all reads: Read name, Base sequence, average quality, sequence length
- Sequence length distribution graph
- Per base sequence quality graph
- Per base GC content graph
- Per base sequence content graph
- Per base N content graph (integrated in the 'Per Base Content' graph)
- Per sequence quality scores graph graph
- Graphs can be expanded to full screen
- All graphs are update in real time as the file is processed
Download link
Version 3.2.3 (released August 2015) can be downloaded here. The size of this program is about 4 MB. No installer needed.
Dereplication is now also available (app). Statistic data about clusters included in Dereplicator.
'Follow' this post to stay up to date.
Requirements:
- <3MB of disk space
- no installation
- no Java
- no .Net
- no admin permissions
- no money :)
Speed & mem footprint:
On an old Toshiba laptop (i5, 2.2GHz) it loads a 0.5GB file in under 11sec (if not processing is applied). This includes also the time for determining the file encoding (Solexa, Illumina, Sanger). The memory footprint should exceeds 15-30MB. I am thinking about doing the file decoding and the data processing in separate threads.
Your feedback
The program was built on feedback from users. So, please comment on things such as:
- Feature requests
- Platform you are interested in (Windows, Mac, Linux) - This is very important!
- Statistics about your files (file type, how many, file size) and your working station (CPU/RAM)
- Which of the already modules are you interested in (so we can improve them)
- New request from users: Allow program resizing so it can fit on very small laptop screens
This tool integrates with Avalanche Workbench.
{kind=link}
Linux, Linux, Linux and Linux. Without the linux support, you are excluding >90% of potential users.
And OS X. Biased sample, maybe, but I just don't see too many folks here with Windows laptops doing informatics work. It's all Linux and OS X for real work.
I agree. There are lots of Mac users in biology field. The port for Linux/Mac is schedule.
Until the Linux port will be available (I promise it will be), the program can be used under Linux via Wine.
Wine is rarely used in bioinfo. For your next project, please take linux/mac as a prerequisite, not an afterthought. Thank you.
we often want to look at one file in a run but almost never would open all files in a sequencing run.
Usually they share many characteristics. Your software should have the option of running as a command line tool as well.
You mean to access the tools via that command line?
yes, like fastqc the program should run from command line if just some non-graphical functionality is needed.
I forgot to mention that it requires that much time only when you open a file for the first time. Opening the file subsequently requires below 1 sec.
it should never need to have the file in memory, except for dereplicate so it should be able to handle files of any size except for that function, correct?
Yes. As you can see in the screenshot the program needs only 38MB for showing a 500MB file.
Cool. Then why this limitation: "On a modest computer (with 3GB RAM) the program should theoretically open files up to 40GB"?
Well, the index in loaded in memory. The more sequences you have, the larger the index. Some calculus shows that it should parse a file with up to 375million sequences, which is equivalent of a 80GB file IF the sequences are about 100 bases each (40GB is for 200 bases/sequence).
Obviously, on a computer with more RAM you could open even larger files. But for the moment the program is 32 bit. The 64 bit version should be ready soon. Then the Linux and Mac versions.
Now I am trying to integrate SFF into the same GUI.
ok but why would you need to load the entire index into memory? after all the user will not actually scroll through hundreds of millions of reads. There is this common flaw, often seen in text editors where opening a large file loads it all up in memory, yet a person only edits or looks at one page at a time.
Update. Now the program will take 15MB of RAM no matter how large the file it is. There will be an update these days available for download.
If you need to perform an operation like getting the viewing, average quality, sorting, etc, you will need to parse all samples. Therefore, you need the index. Probably the index is need for any operation that applies to all samples. Please let me know if you have a different approach. I will try to incorporate it if results in smaller mem footprint.
I think one only need an index if they need to access random parts of the file at high speeds. Reading the all records in an entire file to compute certain values should not need indices.
In your case the only use of an index would be to jump to a read of a given name, this is almost never needed in practice.
I would suggest to make your tool be exceedingly fast and use trivial amounts of memory (I mean megabytes) regardless of the file size. This means a very fast parser and streaming the view rather than preloading it. Now that would be a tool that would separate itself.
Hi Albert!
The technical reasons for keeping the index in memory:
Well I think you should be careful with this and find yourself a collaborator solve their problems. Many things that make sense in theory do not make any difference in practice and do not scale for real work. Good software needs to solve real pain.
It is very rare for example that I would be going back to a fastq file after checking it out once, so the time that it saves me for the second opening is pointless. I just want to look at it once and verify that things look right. For that I would hate to be sitting for minutes staring at a GUI having the memory be consumed when I can do that faster by other means, fastqc will run in the background and I can limit the memory it uses. Moreover it is very likely that the project will have dozens of fastq files associated with it, I would hate having to click and open and wait for each.
So you see how your tool would give me very little reason to use it.
I also have to agree. Normally, nobody would ever look at the fastq-file. Overall statistics are important here (take a look at fastqc). The sequences themselves are not really interesting. They are far too many, anyway. How long can you scroll down your list? ;)
I would recommend you: Talk to biologists or bioinformaticians and ask them, what they really need!
What I would like/need:
These are just three of many many more.... :)
(I agree with Istvan) The reason for an index is random access. The things you are proposing, e.g. viewing parts of a file or doing operations on the entire file do not require an index. For example, if they want to scroll backwards, you can seek().
I edited the original post to show this is rather an editor than a simple viewer (true that for the moment only one editing function is ready :) )
Ok guys. Thanks for feedback. I heard your voices. I will get rid of index :)
Please keep sending good feedback!
Limitation removed!
I would need it to view gzipped files. Our fastq are stored as gzip and dumping them out is a lot of IO hassle. Then let us know when the "linux port" is ready, because the windows machines are kept far away from the valuable data (more IO hassles).
Hi Karl. You mean, you want to work on your packed FastQ file without unpacking the whole file (this may be quite difficult) or you want the program to quietly unpack the program in the background and work on that temporary file? But, yes, I see how this may be a nice feature. Most Linux users probably have their files packed this way.
PS: One possible solution to your packing problem (but works only on Windows) would be not to pack your files with Zip but to use the default packing tool offered by NTFS ("Compress content to save disk space"). A 86MB file gets compressed to 43MB using the NTFS and 25MB using a zip algorithm. Which is not very bad if you consider that 'unpacking' the file is instantaneous.
Have a look at zlib. Reading gzip files is very easy.
Thanks. I was happy to see there is a port for Delphi also. I will take a look.
Update. Now the program will take 15MB of RAM no matter how large the file it is. There will be an update these days available for download.
Adding support for SFF