Entering edit mode

9.0 years ago
Siva
▴
20
Hello guys,
I got 2 files: read1.fastq and read2.fastq, after to have done a mapping with bwa (each reads are 100 b) I've checked the bamtools stats and have this:
***********************************************
Stats for BAM file(s):
***********************************************
Total reads: 820130832
Mapped reads: 812921517 (99.121%)
Forward strand: 418786804 (51.0634%)
Reverse strand: 401344028 (48.9366%)
Failed QC: 0 (0%)
Duplicates: 0 (0%)
Paired-end reads: 820130832 (100%)
'Proper-pairs': 342545373 (41.7672%)
Both pairs mapped: 787752117 (96.052%)
Read 1: 431818871
Read 2: 388311961
Singletons: 25169400 (3.06895%)
Average insert size (absolute value): 309.402
Median insert size (absolute value): 241
So I suppose my reads from illumina hiseq are cleaned, and I got files without adaptors! Am I right?
Regards,
Siva


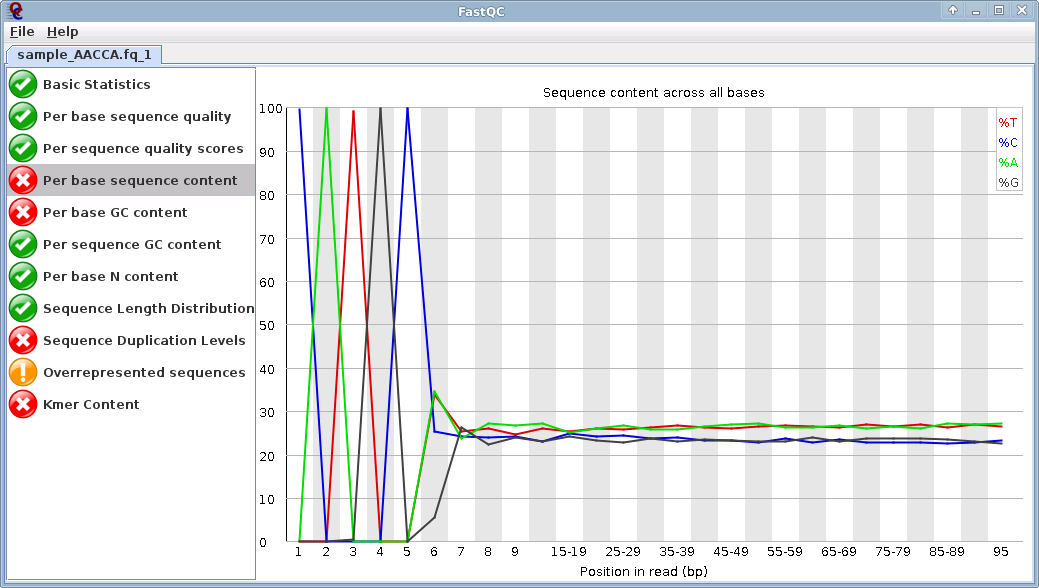
Oh and the mapping is good for the 100 bases, so I really supposed their is no adaptors...