
DNA Methylation Data Analysis
How to use bisulfite-treated sequencing to study DNA methylation
Link: Workshop Website
When? 15 - 17 Dec 2015
Where? iad Pc-Pool, Rosa-Luxemburg-Straße 23, Leipzig, Germany
Scope and Topics
The purpose of this workshop is to get a deeper understanding of the use of bisulfite-treated DNA in order to analyze the epigenetic layer of DNA methylation. Advantages and disadvantages of the so-called 'bisulfite sequencing' and its implications on data analyses will be covered. The participants will be trained to understand bisulfite-treated NGS data, to detect potential problems/errors and finally to implement their own pipelines. After this course they will be able to analyze DNA methylation and create ready-to-publish graphics.
By the end of this workshop the participants will:
- be familiar with the sequencing method of Illumina
- understand how bisulfite sequencing works
- be aware of the mapping problem of bisulfite-treated data
- understand how bisulfite-treated reads are mapped to a reference genome
- be familiar with common data formats and standards
- know relevant tools for data processing
- automate tasks with shell scripting to create reusable data pipelines
- perform basic analyses (call methylated regions, perform basic downstream analyses)
- plot and visualize results (ready-to-publish)
- be able to reuse all analyses
Workshop Structure and Program
This workshop has been redesigned and adapted to the needs of beginners in the field of NGS bioinformatics. The workshop comprises three course modules
Day 1
8 am - 12 am: Introduction to NGS data analysis
- Introduction to Illumina sequencing method from a data analysts view
- Raw sequence files (FASTQ format)
- Preprocessing of raw reads: Idea of adapter clipping and quality trimming
- Mapping output (SAM/BAM format)
1 pm - 5 pm: Linux for bioinformatics
- Introduction to the command line and important commands
- Combining commands by piping and redirection
- Introduction to bioinformatics file formats (e.g. FASTA, BED, VCF, WIG) and databases (e.g. UCSC, ENSEMBL)
- Use of important bioinformatics toolkits (BEDtools, UCSCtools)
- Introduction to R
Days 2 and 3
8 am - 5 pm: DNA Methylation Analysis
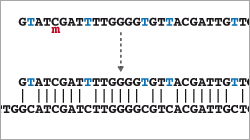
- Introduction to Bisulfite Sequencing
- Read Mapping (special alignment method for bisulfite-treated reads)
- Quality Control
- Data Formats (e.g. vcf, bed, bedgraph, bigwig)
- Overview Statistics
- Tools and Databases (e.g. UCSCtools, BEDtools, UCSC GenomeBrowser)
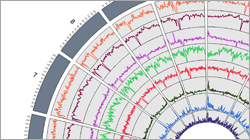
- Visualizing the DNA methylation genome-wide (e.g. Circos Plot, R) or in specific regions/genes (e.g. UCSC, IGV)
- From positions to regions: advantages and disadvantages of segmentation, windowing, and smoothing
- Identification of Differentially Methylated Regions (DMRs)
- Non-CpG Analysis (How to find methylated non-CpGs)
Target Audience
- biologists or data analysts with no or little experience in analyzing bisulfite sequencing data
Requirements
- basic understanding of molecular biology (DNA, RNA, gene expression, PCR, ...)
- the data analysis will partly take place on the linux commandline. Is is therefore beneficial to be familiar with the command line and in particular the commands covered in the Learning the Shell Tutorial
Included in the Course
- Course materials
- Catering
- Conference Dinner
Trainers
- Helene Kretzmer (University of Leipzig) is working on DNA methylation analyses using high-throughput sequencing since 2011. She is responsible for the bioinformatic analysis of MMML-Seq study of the International Cancer Genome Consortium (ICGC).
- Dr. Christian Otto (CCR-BioIT) is one of the developers of the bisulfite read mapping tool segemehl and is an expert on implementing efficient algorithms for HTS data analyses.
- Dr. David Langenberger (ecSeq Bioinformatics) started working with small non-coding RNAs in 2006. Since 2009 he uses HTS technolgies to investigate these short regulatory RNAs as well as other targets. He has been part of several large HTS projects, for example the International Cancer Genome Consortium (ICGC).
- Dr. Mario Fasold (ecSeq Bioinformatics) has developed several bioinformatics tools such as the Bioconductor package AffyRNADegradation and the Larpack program package. Since 2011 he is specialized in the field of HTS data analysis and helped analysing sequecing data of several large consortium projects.
Key Dates
- Opening Date of Registration: 1 June 2015
- Closing Date of Registration: 15 November 2015
- Workshop: 15 - 17 December 2015 (8 am - 5 pm)
Attendance
- Location: iad Pc-Pool, Rosa-Luxemburg-Straße 23, Leipzig, Germany
- Language: English
- Available seats: 24 (first-come, first-served)
Registration fees: 998 EUR (without VAT)
Travel expenses and accommodation are not covered by the registration fee.



About Leipzig
Leipzig is a modern city with many students, an international flair and an established cultural scene. There many parks and an exciting night life. Leipzig features one of the largest and most beautiful christmas markets in Germany which takes place during the workshop. Take a look.
Leipzig is only about one hour away from Berlin and three hours away from Prague. It can be conveniently reached by car, bus, train or plane (for example via Leipzig/Halle Airport or one of the Berlin airports). Find more information about Leipzig on its official webpage.







Dear Lars,
we will provide the machines. Every participant will get his/her own computer. You don't have to bring your laptop!