Hi Tonja,
The rationale for the use of a binomial test is described in the paper: http://www.ncbi.nlm.nih.gov/pubmed/20436461
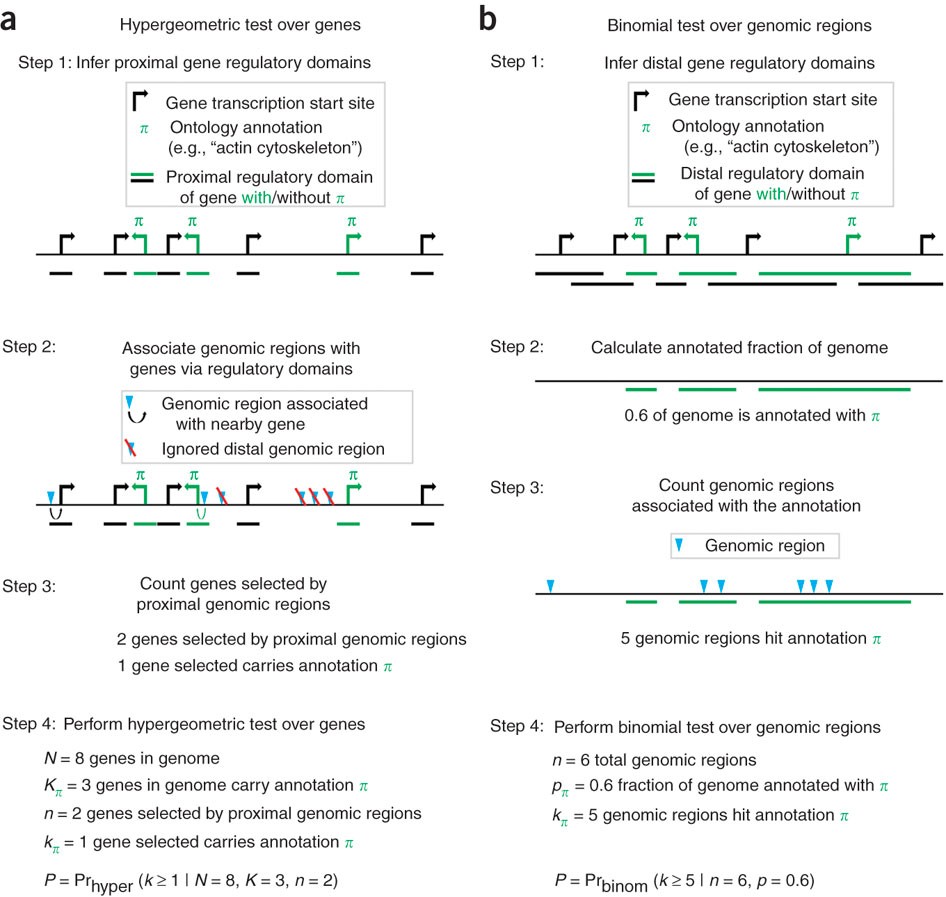
In the introduction, in the second paragraph, they say "...the standard approach to capturing distal events--associating each binding site with the one or two nearest genes, introduces a strong bias toward genes that are flanked by large intergenic regions' and then they further explain that this bias leads to the generation of false positive enrichment scores.
The hypergeometric test is not biased in this way for PROXIMAL regions because they do not have such wide variation in size (the authors state they are between 2-5kb). But the same cannot be said for DISTAL regions - they have huge variation in size.
So, to avoid this enrichment, the hypergeometric test is not used...Think of it this way, if you used the same procedure in Figure 1a as you did in Figure 1b (i.e., hypergeometric test both times), then genes that had huge flanking regions would pop up in your analysis way more than genes with shorter flanking regions, due to probabilistic considerations introduced by the sheer size of the region.
So, instead they define a regulatory domain and the number of bases that that covers (step 2 in Figure 1b), then convert to a fraction (see Results, page 495-496). This is a much better approach than using the hypergeometric test because it is free from the type of bias they describe.
In answer to your question, yes, you can use a hypergeometric test any time you wish to test for enrichment of items. However, in this case, applying it is likely to lead to a biased (systematically inaccurately estimated) test statistic.
If you still have questions after reading this, reading the paper plus references 12,15, and 16 should clarify the issue beyond and doubt.


Hi Tonja, I am sorry, but the sentence, "Somehow I do understand why it is not possible to extend the regulatory domain (the way they do it in the paper) and apply a hypergeometric test on those domain?" is unclear. Do you mean you do not?
I corrected it