
Here's the situation.
I have proteome files for a bunch of strains. Each strain has its own fasta proteome (strain1.faa, strain2.faa, strain3.faa).
I also have a fasta list of AA sequences, and I want to know if they are present within these strains. That "query" file, looks like this:
>gene 1
MKGMF...*
>gene 2
MQWAEA...*
etc...
What I want in the end is a matrix with the strains in first column, and first row being the genes. I DONT want to have to do a manual blast for every cell because that's impractical. I just want the information. The values in the matrix is the %identity of that gene in that strain. It will look like this:
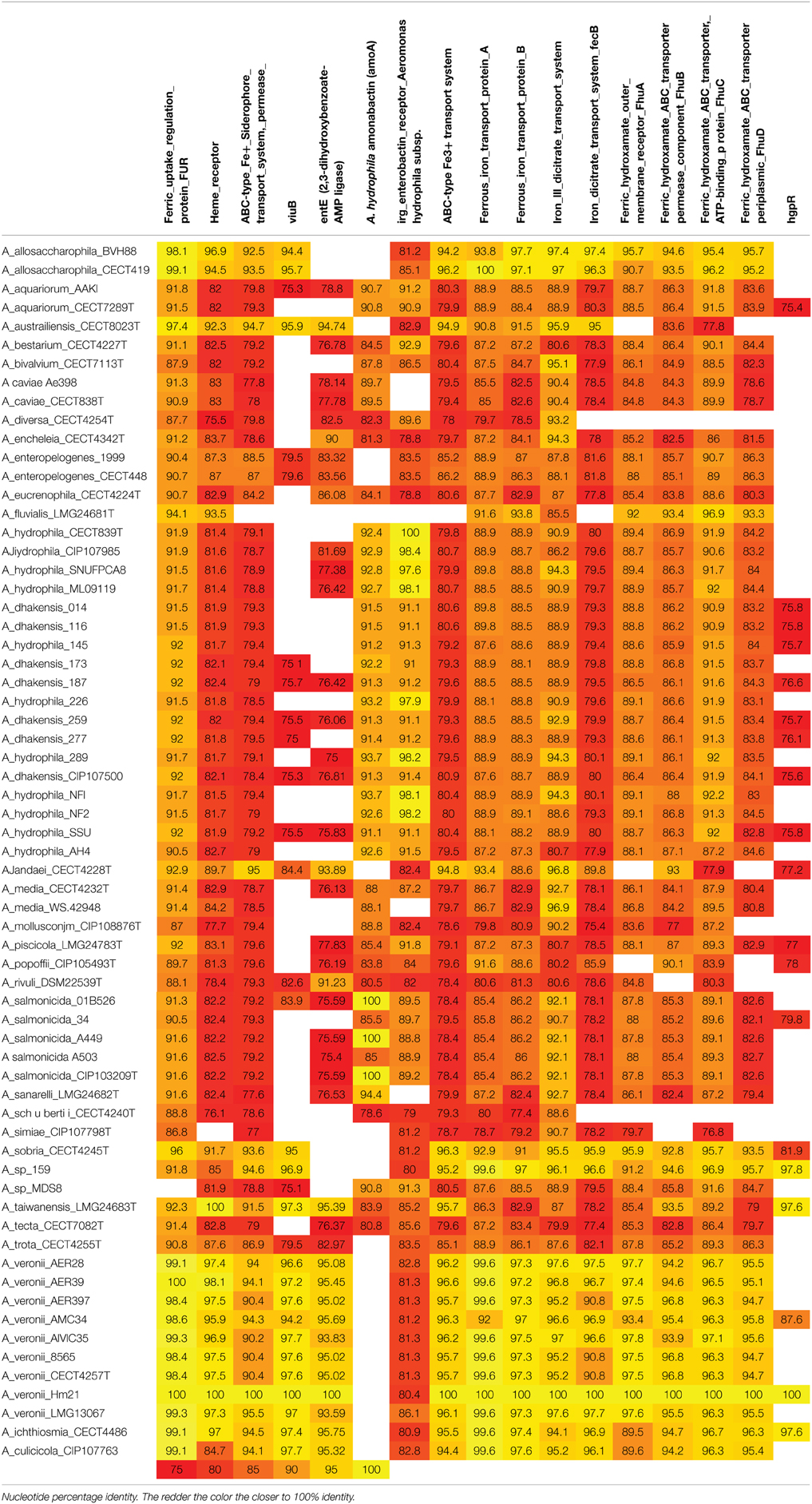 What is the most parsimonious way to go about this project?
I have a lot of strains, and hundreds of genes to test. But, I'm okay with outputing a csv for now. It's such a large task that I'm unsure of how to start it.
What is the most parsimonious way to go about this project?
I have a lot of strains, and hundreds of genes to test. But, I'm okay with outputing a csv for now. It's such a large task that I'm unsure of how to start it.


It's not the heatmap I want. It's just the raw information. I don't want to have to individually do a blast search manually for each cell.
I can't find another google image picture that depicts this very type of project.