Hi I am trying to compare the effects of 2 transcription factors.
I have the initial data files in .bed format on which I used MACS to get output .bed files again for peak calling.
My genome does not have a specific GTF file and it is not fully annotated so I only have a GFF3 file.
AnnotatePeaks.pl of HOMER worked well and I have the TSS binding sites of genes isolated.
My question is
I want to plot a line plot from -1000 to +500 relative to the ATG region of each binding sequence to show the average binding signal over all genes for both the transcription factors.
Most of the programs I saw use GTF files which I do not have!
Also is there is a way to combine both the transcription factor peaks together on a single plot? I couldnt really find a tutorial that puts the TSS at ATG at position 0 and makes a graph.
Converting gff3 to gtf should not remain a problem. Many tools/scripts are suggested in the forum. Please see if the below method that I usually follow would help you with it.
If you understand R code, this is my code for doing exactly what you want. I wrote this a while ago so my code is awful but you might be able to pull out some useful stuff! http://rpubs.com/achitsaz/94710
I have written R script which can probably solve your problem. For given set of genes in bed format, it will give you averaged line plot or even you can generate heatmap for each gene. Script needs two files
1) set of genes in bed format
2) alignement data in .bedgraph format.
That is exactly what I was looking for although I don't know the format of how your diffReps_output_gene1_X1.txt looks like so that I can similarly modify my own files.
I did not find what I was looking for in SeqMonk. Anyone knows if Seqmonk can do this?
Looks like you figured it out but just in case: Sorry, should have been a little more clear. diffreps is a differential binding analysis software. The diffreps.txt file (output format here) was used to subset the genes that I wanted to look at that were called different using differeps. I took the genes from the differeps output that were mapped 500 basepairs from a TSS and have a log2FC > 1.5. If you look at code lines 12 and 13, I then took those genes and pulled them out of my GRanges transcriptome, which I then used for analysis.
Is there any way I can isolate reads per a specific bin? For example between '-500 to 0' or between '-1000 to -500' ?
Also can I change the size of the bin?
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000))
Can I change this interval i.e. the size of the bin? Something like axis(side=25, at=c(0:1000))







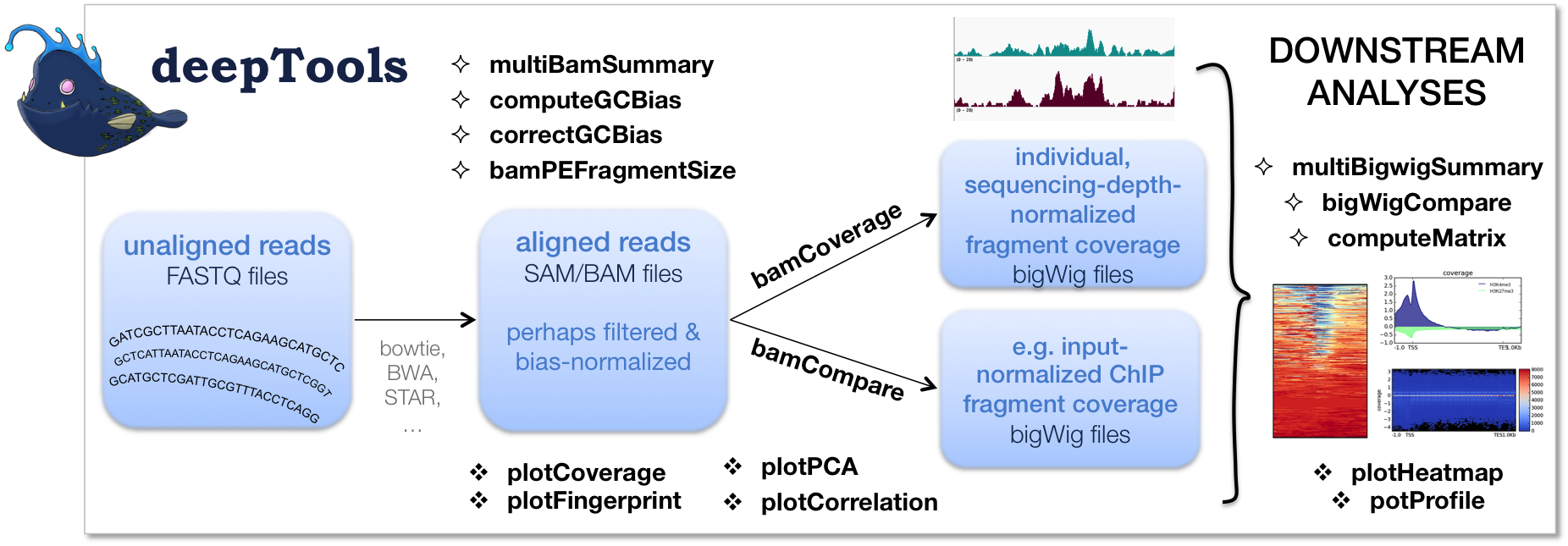
Converting gff3 to gtf should not remain a problem. Many tools/scripts are suggested in the forum. Please see if the below method that I usually follow would help you with it.
gff3ToGenePred yourgenemodel.gff yourgenemodel.genePred
genePredToGtf file yourgenemodel.genePred yourgenemodel.gtf
Link for gff3ToGenePred and genePredToGtf : link
I am not sure if this aligns with what you are trying to do. You can try using https://github.com/shenlab-sinai/ngsplot