
Hi all,
I'd like to introduce you a cross-platform and every fast FASTA/Q toolkit, Seqkit, written in Golang.
- Documents: http://bioinf.shenwei.me/seqkit (Usage, FAQ (New!), Tutorial, Benchmark and Development Notes)
- Source code: https://github.com/shenwei356/seqkit[![GitHub stars]6]


Latest version:





Citation:

Introduction
Common manipulations of FASTA/Q file include converting, searching, filtering, deduplication, splitting, shuffling, and sampling. Existing tools only implement some of these manipulations, and not particularly efficiently, and some are only available for certain operating systems. Furthermore, the complicated installation process of required packages and running environments can render these programs less user friendly.
SeqKit provides executable binary files for all major operating systems, including Windows, Linux, and Mac OS X, and can be directly used without any dependencies or pre-configurations. SeqKit demonstrates competitive performance in execution time and memory usage compared to similar tools. The efficiency and usability of SeqKit enable researchers to rapidly accomplish common FASTA/Q file manipulations.
I had used SeqKit to solved some problems raised by Biostars users in simple and efficient ways. For examples:
- How to get contigs from scaffolds
- parsing fasta file
- How to append strings (from one file) to Fasta headers (in another file)
- Renaming fasta file according to a name list (blast output)
- Filter Fasta using regexp on header
Benchmarks
SeqKit uses author's lightweight and high-performance bioinformatics packages bio for FASTA/Q parsing, which has high performance close to the famous C lib klib (kseq.h).

FASTA manipulations
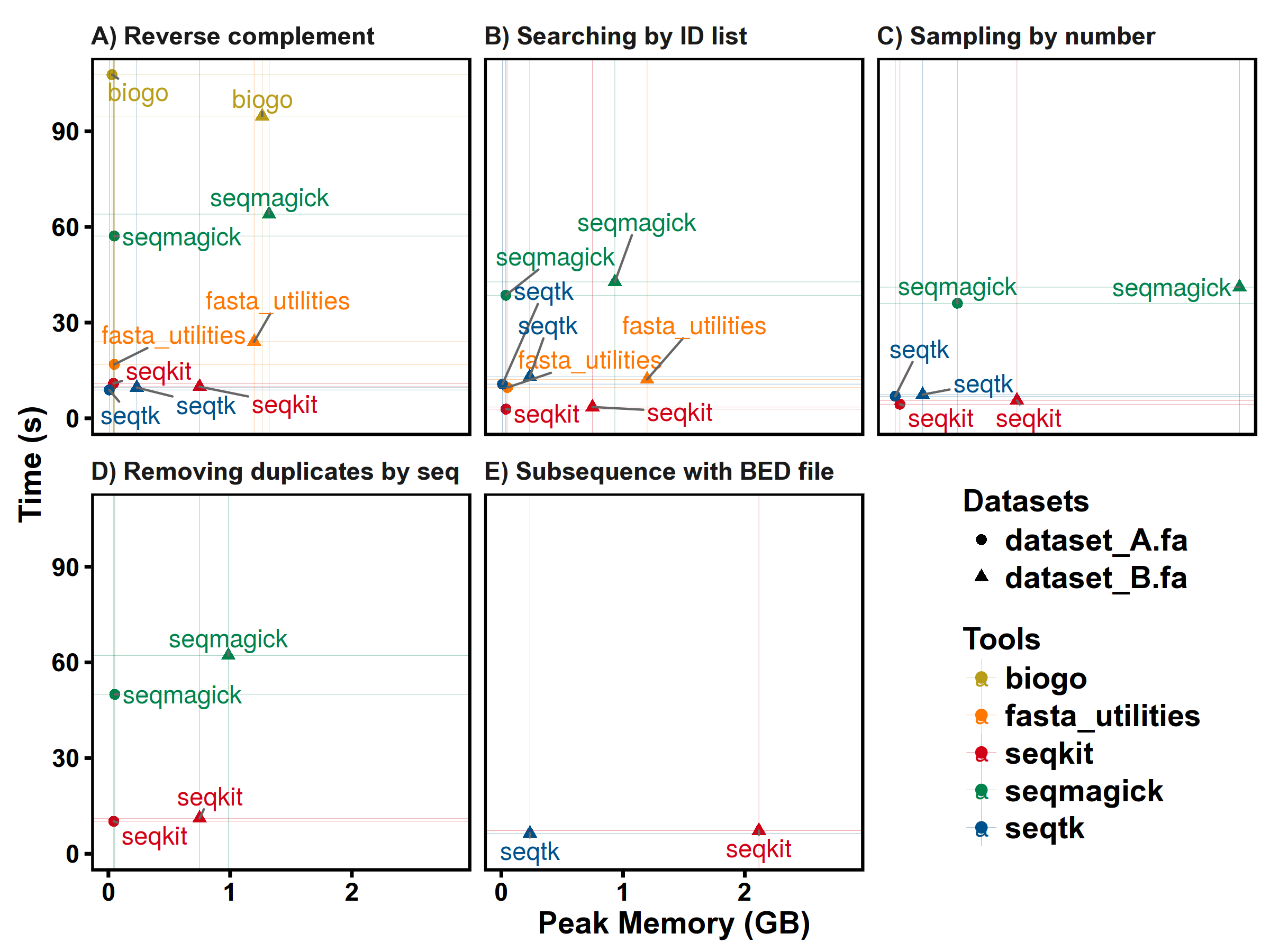
FASTQ manipulations
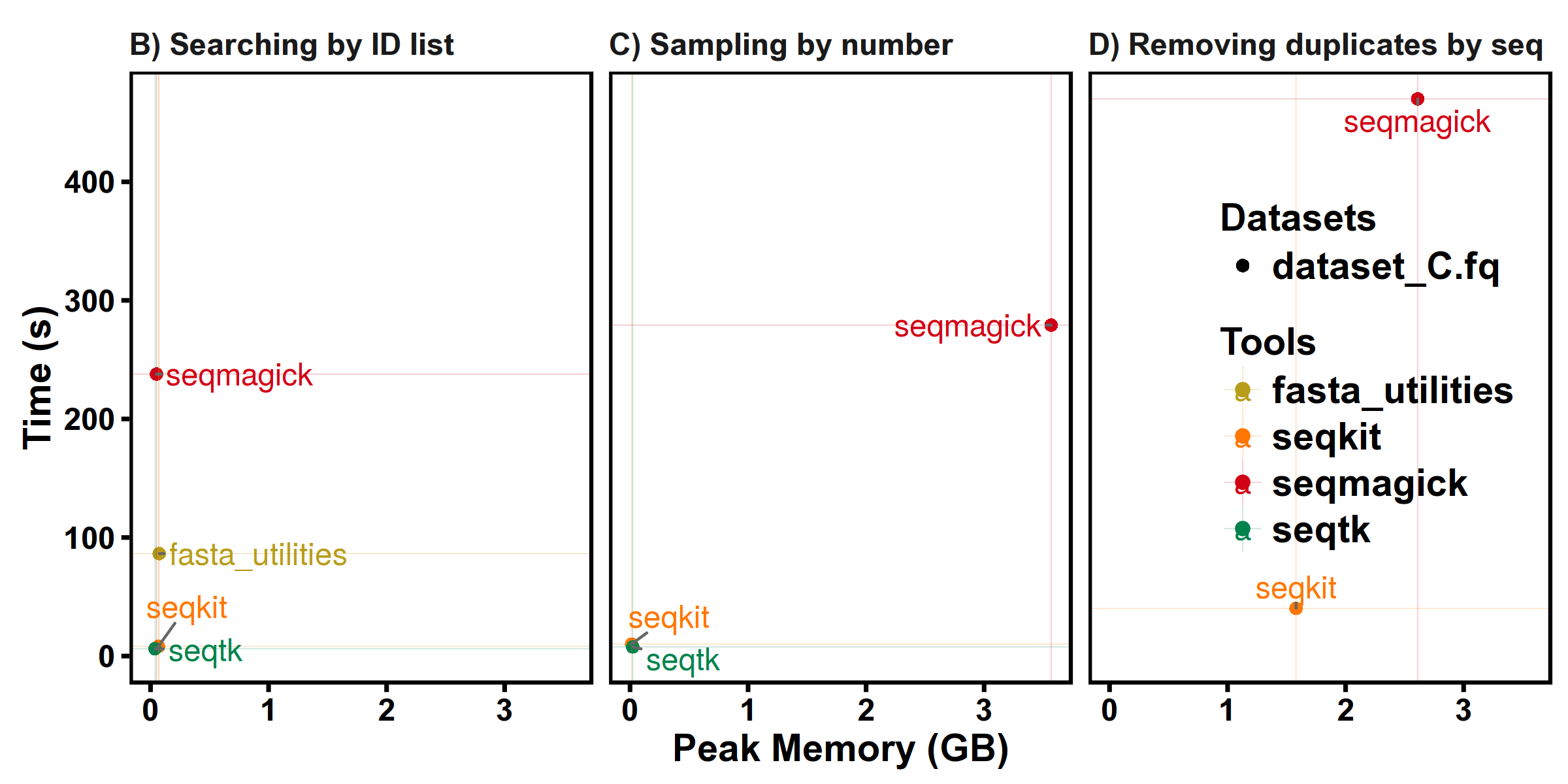
Subcommands
Sequence and subsequence
seqtransform sequences (revserse, complement, extract ID...)subseqget subsequences by region/gtf/bed, including flanking sequencesslidingsliding sequences, circular genome supportedstatsimple statistics of FASTA filesfaidxcreate FASTA index file
Format conversion
fx2tabcovert FASTA/Q to tabular format (and length/GC content/GC skew)tab2fxcovert tabular format to FASTA/Q formatfq2facovert FASTQ to FASTA
Searching
grepsearch sequences by pattern(s) of name or sequence motifslocatelocate subsequences/motifs
Set operations
rmdupremove duplicated sequences by id/name/sequencecommonfind common sequences of multiple files by id/name/sequencesplitsplit sequences into files by id/seq region/size/partssamplesample sequences by number or proportionheadprint first N FASTA/Q records
Edit
replacereplace name/sequence by regular expressionrenamerename duplicated IDs
Ordering
shuffleshuffle sequencessortsort sequences by id/name/sequence
Misc
versionprint version information and check for update


I just used seqkit to make a shell wrapper to take fasta length distribution that I wanted to share in order to let you know that how useful this (seqkit) could be.
Script
Output
Ofcourse, there is scope for improvement and can be modified according to requirements. For me, that was required!
Thank you my friend Wei
How about outputting sequence lengths and ploting using other tools
Or
That's even better !!
Hi,
I am trying to extract sequences from a gzipped fastq file(17GB) using sequence ID list in a text file (2.8GB) using the following:
seqkit grep --pattern-file id.txt raw-reads.fastq.gz > subset.fastq.gz
However, the resulting subset.fastq.gz file is empty. Could you please tell how to deal with such huge files? Or is the command is incorrect in the first place?
Can you post the output of
head -6 id.txt?head -6 id.txt
@D00723:299:CCRTLANXX:1:1101:1281:1987 2:N:0:1
@D00723:299:CCRTLANXX:1:1101:1301:1993 2:N:0:1
@D00723:299:CCRTLANXX:1:1101:1660:1986 2:N:0:1
@D00723:299:CCRTLANXX:1:1101:1769:1980 2:N:0:1
@D00723:299:CCRTLANXX:1:1101:1755:1982 2:N:0:1
@D00723:299:CCRTLANXX:1:1101:2165:1989 2:N:0:1
you need remove the leading symbol
@byHello, I am trying to transform some fastq.gz files to their reverse complement, but when I run the command
I get the output printing to the console. Is that what this function is supposed to do? I want to edit the file itself.
Thank you
Reposting because I accidentally posted as a reply: I am trying to transform some fastq.gz files to their reverse complement, but when I run the command
I get the output printing to the console. Is that what this function is supposed to do? I want to edit the file itself.
Thank you
Why would you want to edit the input file itself? Most programs will not allow you to edit the input file with output. Why not write the results to a new file and use that?
I found out my reads were the reverse complement of what I needed to run my analysis (in a program downstream of this step). I did send the output to a new file (with >), which messed up the fastq format, but did have the reverse complements.
Can you try running
seqkit seq -rp hairpin.fastq.gz > rev_comp.fastq? You should provide program options before input and output files. I just tested this out and encountered no fastq corruption.You can also use
reformat.shfrom BBMap suite to do this.Thank you @GenoMax! This helps tremendously! The seqkit command worked perfectly!