
Hi all,
I've come across a weird issue with my RNA-seq data.
Some details about the data:
There 4 libraries of RNA, all total RNA-seq in wild-type and mutant cell with 2 replicate. Total 2 wild-type(Lane1, Lan2) + 2 mutant(Lane3, Lane4).
- Platform: Ilumina Truseq
- Prep kit: TruSeq LT Kits and TruSeq v1/v2 Kits
- Adaptor: Lane1--TruSeq Adapter, Index 4; Lane2--TruSeq Adapter, Index 6; Lane3--TruSeq Adapter, Index 12; Lane4--TruSeq Adapter, Index 12.
After I got the RNA-seq data, I ran FastQC.
In the result I found:
- Failed Per base sequence content --- might due to random hexamer priming bias (Commonly seen in RNA-seq data)
- Failed Kmer content --- Both in R1 and R2. The weird thing is that the over-represented Kmer are perfectly consistent at the 5' of R1. There is a perfect 12 bp sequence that at 5' of R1 :
CAGACCGCGTTC. This only appears on R1 not in R2, and it is all across 4 libraries. Although R2 also has kmer at 5', their order is not consistent and the peak is somewhat random. This how it looks like:
Lane1-R1

Lane1-R1

Lane2-R1
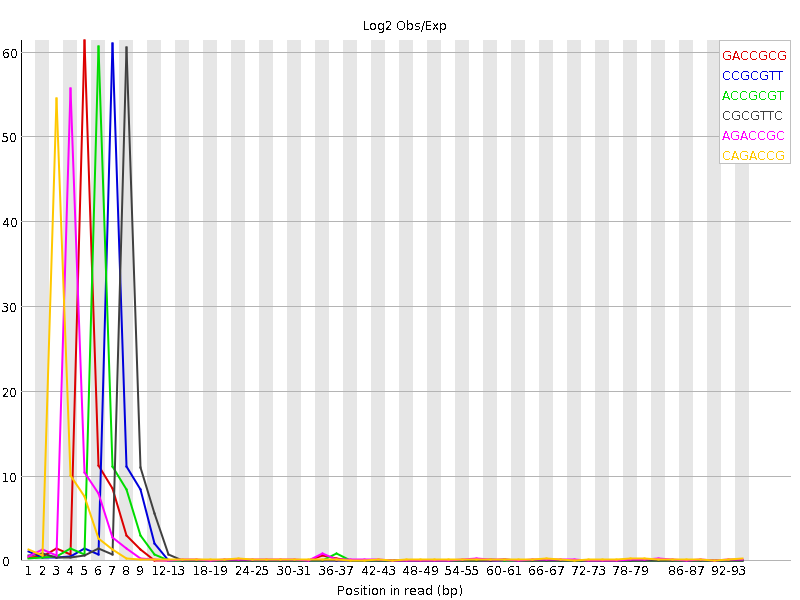
What I've done:
- Blast --- no result
- Check the adaptor sequence --- no match
Here is the all the image: https://goo.gl/photos/UGqMseNycR99vhVu9 (I cannot upload my image to free image hosting website. My images are saved via right click save as from the browser. This is weird.)
Similar issue(Despite that he managed to find the source of the kmer): http://seqanswers.com/forums/showthread.php?t=45539
Anyone has any idea about what is going wrong here? Thank you so much for you input!
There is probably nothing wrong. This may represent signature pattern seen at the beginning of RNAseq reads. You should read this blog post by Dr. Simon Andrews, author of FastQC.
It's spelled 'weird' (sorry, but it drives me nuts...)
LOL. Noticed that already. Thanks for the correction!
There's at least one more instance in your post ;P