Entering edit mode

6.9 years ago
EagleEye
7.5k
Hello everyone,
I have ChIP-seq samples with replicates for the same experiment. I have processed ChIP-seq as follows,
Steps done:
- Bowtie and Bowtie2 (50 bp read length, used both outputs for further processing)
- picard (duplicate removal)
- NGSUtils (BAMutils, clean with Black list and MAPQ 30)
- Deeptools (fingerprint, BAMsummary and correlation)
Query:
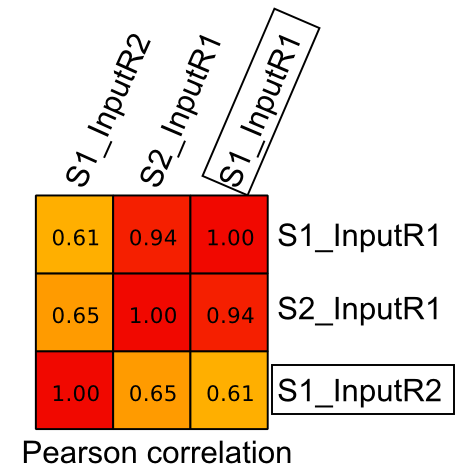
I am planning to do peak calling using this replicate samples (2 x treatment and 2 x Input) with MACS2. But when I check the correlation (pearson) between Input replicate samples (S1), it gives 0.61 (highlighted with box) unlike in treatment replicates it was above 0.85 [refer attached figure below].
Is it fine to consider S1_InputR1 and S1_InputR2 as replicates when doing peak calling with MACS2 ? [refer attached figure below]
Please give some suggestion.

What does the Spearman's correlation look like?
With Spearman correlation it got reduced to 0.44 for Input samples (S1_InputR1 vs S1_InputR2) but not for treatment samples (0.84).
Interesting, do the fingerprints also look different?
Here is the fingerprint for above samples,
The Inputs prepared during two different experiments looks much closer (S1_InputR1 and S2_InputR1) than the Inputs from the same experiments (S1_InputR1 and S1_InputR2).
Interesting, S1_InputR2 also covers ~5% less of the genome. From the graph, it looks like the depth might be a bit lower for it, but I'm guessing it's not hugely different. If you have a recent-ish version of deepTools, it'd be interesting to use
--outQualityMetrics something.txt --JSDsample S1_InputR1.bamto see what the "synthetic JSD" is. This is the Jensen-Shannon divergence between a sample's coverage distribution and what would be seen from an ideal input sample sequenced to the same depth. Lower is better and I suspect your S1_InputR2 will have a notably higher value, which indicates that something went wrong at some point (possibly too many PCR cycles?) and maybe the sample should be excluded.