Entering edit mode

6.8 years ago
anu014
▴
190
Hi Biostars !
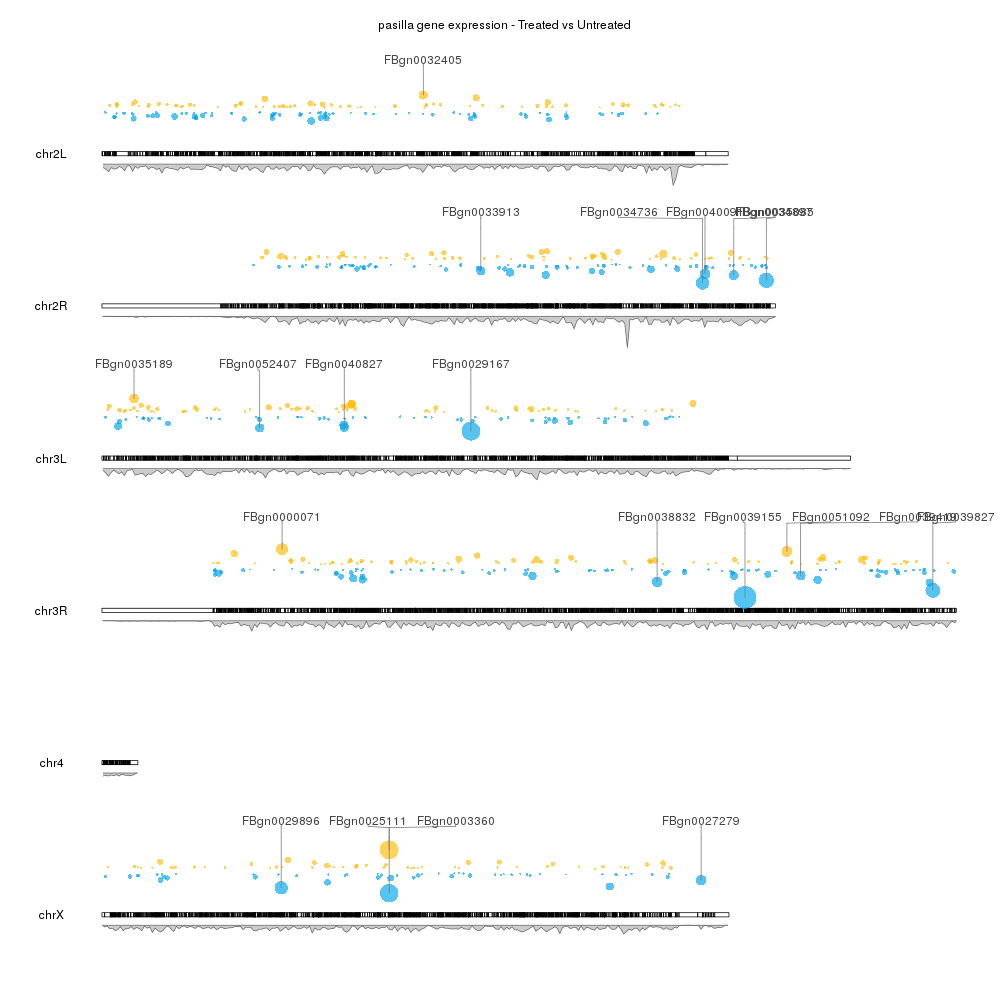
Can anyone tell me how can I plot up-regulated & down-regulated genes across the genome such that the y-axis is fold change & x-axis is position of genes on genome?
Thank you in advance :)
Good Day!



You can find an example that might be helpful in karyoploteR's Examples and Tutorial site. It has complete code to get to something very similar to what you asked for starting from a DESeq2 analysis (the example is with a D. melanogaster dataset, but it would work the same with any other organism)