Entering edit mode

6.2 years ago
arronar
▴
280
Hello.
I'm interested in creating adjacency matrix for genes-coexpression network from a microarray data and I would like to know which methods you guys are using for such a procedure.
Till now, I figured that there is the WGCNA but I don't know if it can calculate the adjacency matrix and also there is a package called coexnet that support that can calculate the adjacency matrix but nobody is referring to it and I cannot understand if it's robust or not.

Thank you very much for that extended list. After reading about them, I decided to use the WGCNA and more accurately its function called
bicor(). Now let me ask you something about the procedure. So far on my microarray experiment, I ran RMA algorithm, completed the annotation step and divide the matrix into 3 separate matrices, one for each sample. The initial one was[3xControl, 3xTreatment1, 3xTreatment2]while now I have one matrix with3xControl, one with3xTreatment1and one with3xTreatmen2.What I want to do as I said in my I.P is to calculate the adjacency matrix (with beta = 1) for each one of them. My question now is if there is a need for summarizing somehow the 3 repetition samples in each condition. Should I calculate the mean or something else - like MAD - for each gene (row) for the same conditions?
I have not used
bicor()- sorry. I also believe that there are more flexible network methods than WGCNA, despite the enormous popularity of this program. This said, I do not believe that you have to summarise your replicate (repetition) samples. If you want to summarse them, however, then summarise them by the mean value per gene.@Kevin this is the kind of analysis which i was looking for as i kind of not sure how to go with differential expression between various stages ,the go for network analysis and show the differences or similarity , "selected from proteins with degree > 10, BC > 0.05 and CC > 0.3" so for this did you just discard those nodes which were below your prescribed threshold ? and then form a network
Second one "titin network with first neighbourhood was constructed using titin as seed in the two " so you constructed this one from Biogrid ?as a reference network ,"titin network was then merged with the TMD network using " ? this is something im looking into what kind of similarities or differences that comes with each stage of differentiation ,but im not sure how to proceed ,so far im using WGCNA but your approach is also another of doing...
Dear krushnach,
With regard to the parameters to filter the key nodes of the network, they come according to the number of nodes and vertices of the network. If you have a large network, you put a high degree, BC and the CC, and the same if it is a small network, you can put a smaller cut-off.
For the construction of the titin network, We used STRING v 10.5 database (Szklarczyk et al., 2015), Genemania (Montojo et al., 2014) and BioGRID v. 3.4 (Chatr-aryamontri et al., 2017), as it was just one seed, it was easier to use several databases.
And then all the files generated in those databases were exported to Cytoscape and this allows the merger of two or more networks.
Tools → Merge → Networks....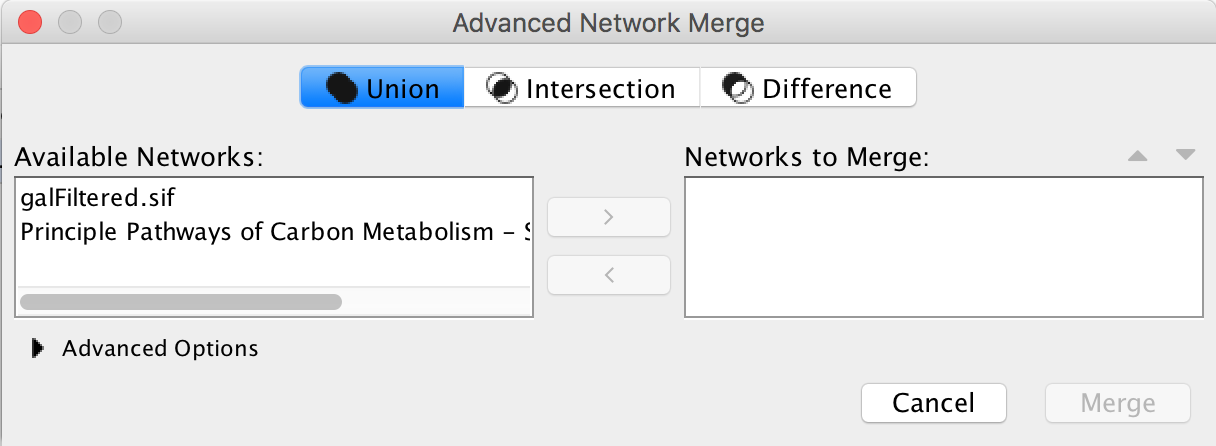
As you can see here.
Best regards,
Leite
Hello krushnach. Regarding the degree, BC, and CC, these were just used to identify key nodes in the pre-existing network. There was no new network formed after this. By identifying the key nodes, one can then build a 'discussion' and 'hypothesis' around these key nodes.
For the other part on the titin network, let me see if I can get help from someone (I will ask them to reply here).
Hello @kevin yes for titin " titin network" in the paper you mentioned "titin" as seed sequence "aforementioned interaction databases as well as the BioGRID v. 3.4 " so im not clear about the seed sequence ? so as an input you gave just that single gene to BioGrid and found out the interactions ?
Dear krushnach80,
In our study, we did not use the "TTN" sequence as seed but rather the gene itself. And yes, just put the name of the gene of interest in BIOGRID as well as in the other databases and find the interactions.
" "TTN" sequence a" sorry for the mistake since i didn't have the concept of seed in networks so that error happened ..