
we have a pilot project consisting of one WGS paired-end sample from mouse. The mouse were inserted with a transgene of known sequence. We would like to identify possible insertion sites for this transgene.
I was wondering which tool can help me do that.
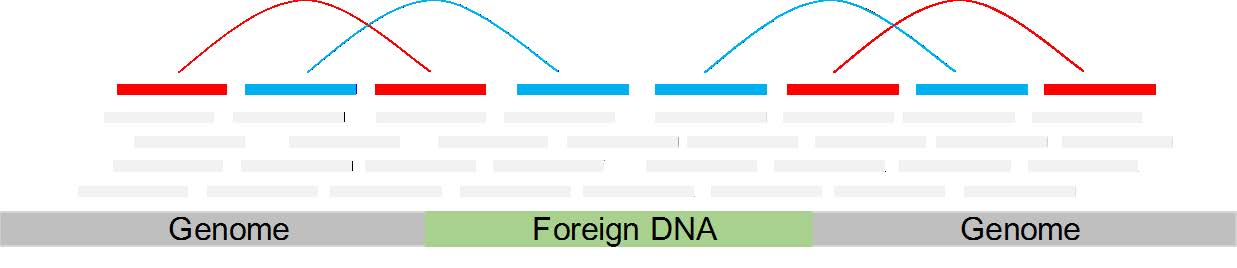
What I was thinking to do is to map the reads to the mouse genome (using soft-clip and no mismatches allowed) and than look for reads which are splitted. This reads might be splitted, because only part of them is mapped to the mouse genome, while the other half would map to the inserted transgene (s. image - these would be the red-colored reads in the middle).
Another method would be to look for pairs with an insertion site larger than expected which might happens because the two reads spann the insertion site.
A third method would be to look for orphan reads, extract them and their mates and see if it is belong to the transgene (the blue-colored reads in the image).
While I know some tools to try and do the second option (e.g. DELLY), I am not really sure if the first and third options are not better and more precise. Unfortunately I 'm not sure how to do these.
I would appreciate any ideas to do any of the above. Are there any tools for split-read analysis of this kind or to identify orphan reads?
Thanks in advance
Assa


This past thread may be useful: Identification of the sequence insertion site in the genome