Entering edit mode

6.0 years ago
williamsbrian5064
▴
510
Hi,
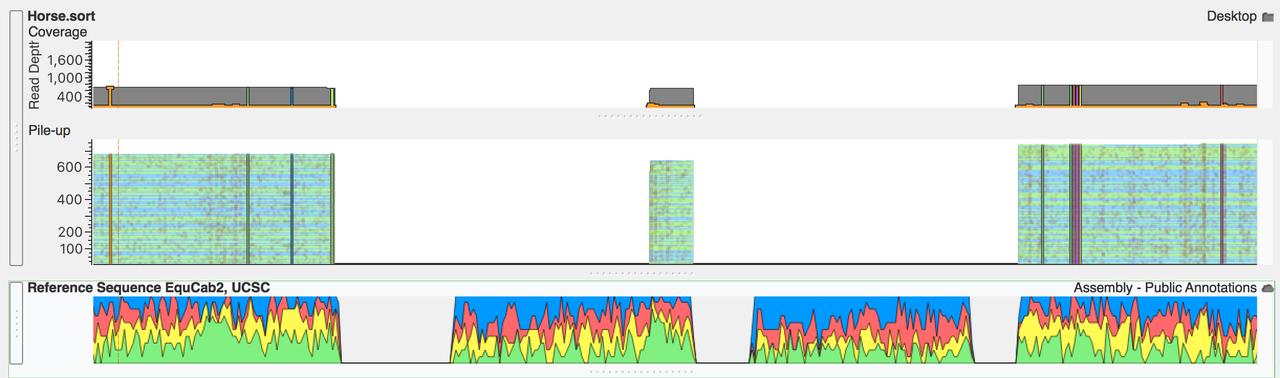
I have posted about this before and I got great responses! I was directed to use a program called Minimap2 to align Pacbio reads. We sequenced an area that has a few large gaps in the reference genome. We are particularly interested in what lies in the gaps in the reference. My issue is that I am struggling to get the reads to map over the gap. Does any one have any suggestions how to get the reads to map over the gap using MiniMaps2?
We did sequence some controls. Maybe I should just insert the known sequence into the reference fasta and reindex the fasta file? Would that work?
The sequencing did sequence the unknown areas in the reference so it isn't an issue of failed experiment.
Thanks for the help
Reminds me of this recent issue: https://github.com/lh3/minimap2/issues/155
I will check it out. Here is a picture of what that output bam looks like if anyone is interested. I the fastq file only contains reads that cover the entire gap. I think there about 300 reads. I have no idea why its cutting up the data like that. None of the reads are that short.