Entering edit mode

5.9 years ago
Hi everyone, I'm using trimGalore to remove adaptors and trim low quality reads.
The problem is that I get almost the same output after quality control as before it.
Can anyone tell what's the problem ?
This the command I used
$ trim_galore --fastqc --illumina 23_S19_L001_R2_001.fastq.gz -q
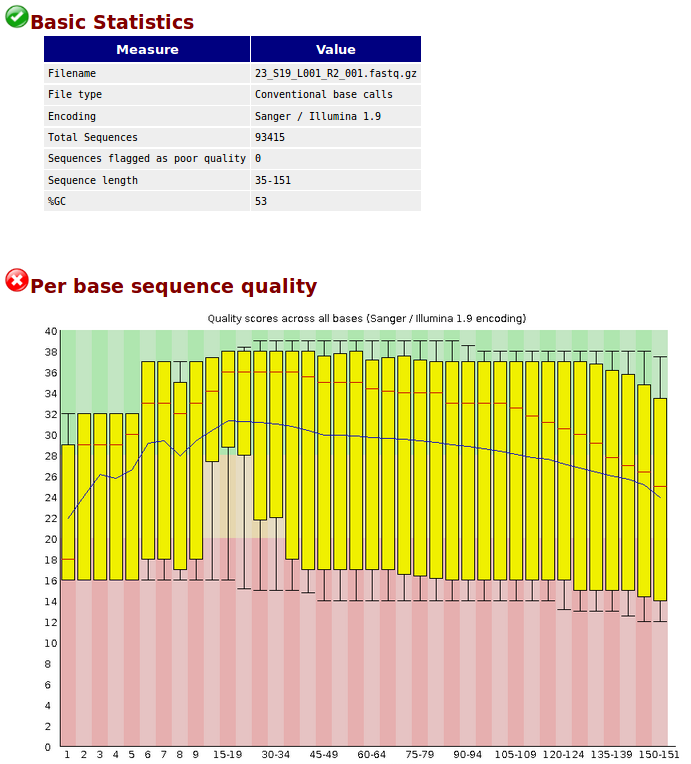
before quality control :https://s9.postimg.cc/7aku795yn/before.png
{kind=link}
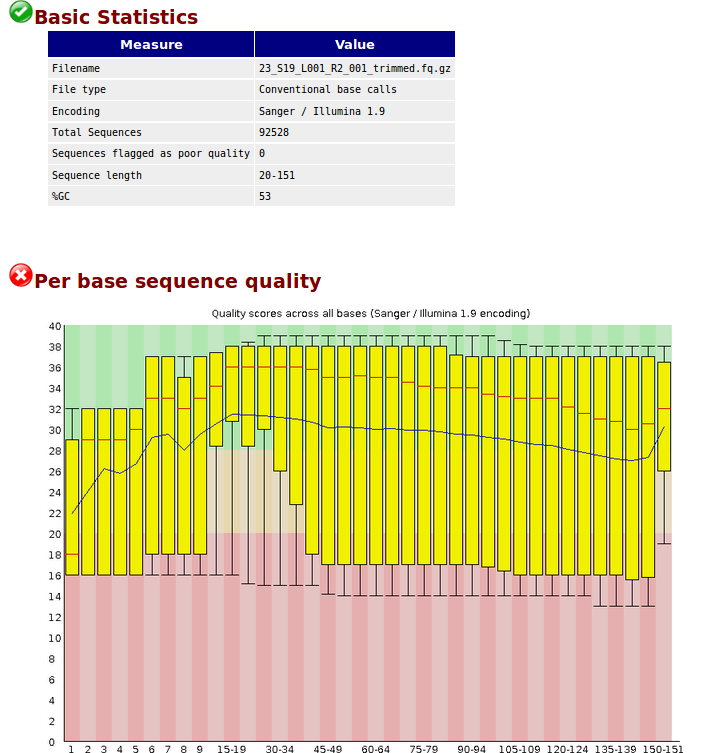
after quality control :https://s9.postimg.cc/93nqvaohr/after.png
{kind=link}
Strange that it worked for you without specifying quality
INTin-qoption. It failed in my caseI checked the help
Can you check once again?
UPDATE
Specifying
-qworks and only towards end of the reads. But I need to confirm.Are you sure you need to remove adapters? That may already have been done by the sequencing machine (in which case you probably won't see much difference in QC)
How to add images to a Biostars post
The result of the trimming looks ok for me.
The quality of your data set is not the best. Removing adaptors and trimming low quality ends from reads (-q) leads to an increase in the overall mean quality values towards the end, which is visible in after.png. Given the before.png result, I would not expect too much.