
Hello Biostars community,
I'm trying to analyze miRNASeq data of TCGA melanoma (SKCM) samples. This is a prototype analysis in which I picked hsa-mir-155 and looked at how its expression is correlated with survival. My end goal is expanding this analysis to other cancer types also focusing on other miRNAs.
I performed my analyses with two of the popular packages in R (RTCGA and TCGAbiolinks) and obtained quite different results. I'm trying to make sense of what might be causing the different results. Any help is appreciated. For comparison purposes, I'm attaching some figures and my codes as well.
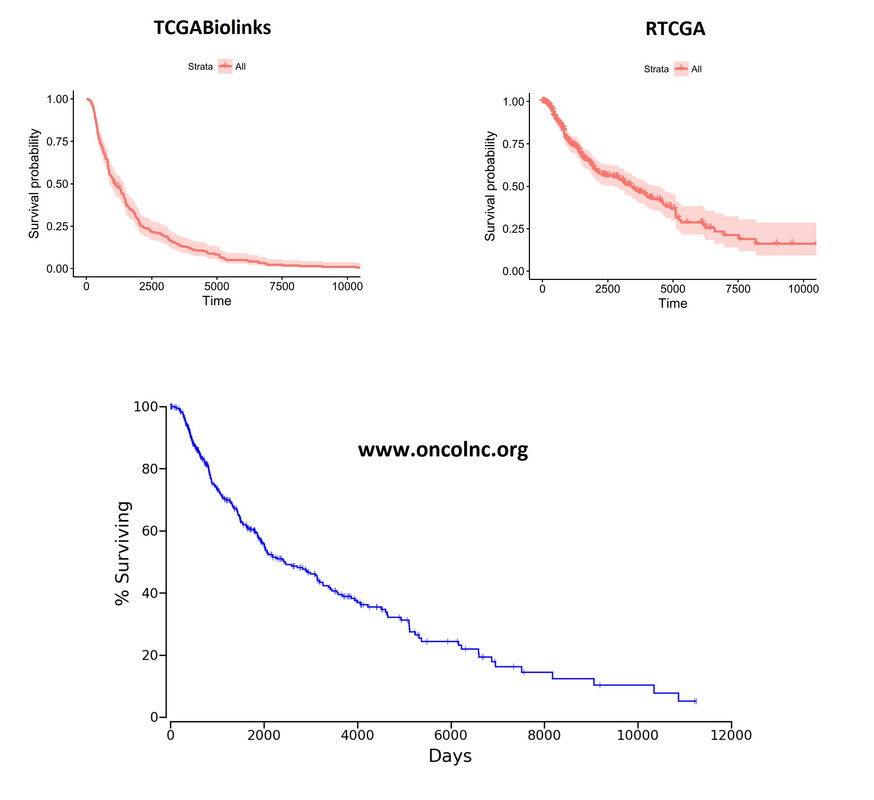 Overall Kaplan-Meier curves look quite different. These plots were generated on the whole cohort (unsegregated for any parameter). I'm including the plot from
Overall Kaplan-Meier curves look quite different. These plots were generated on the whole cohort (unsegregated for any parameter). I'm including the plot from oncoLNC.org database as well for comparison purposes. RTCGA looks more like the oncoLNC curve.
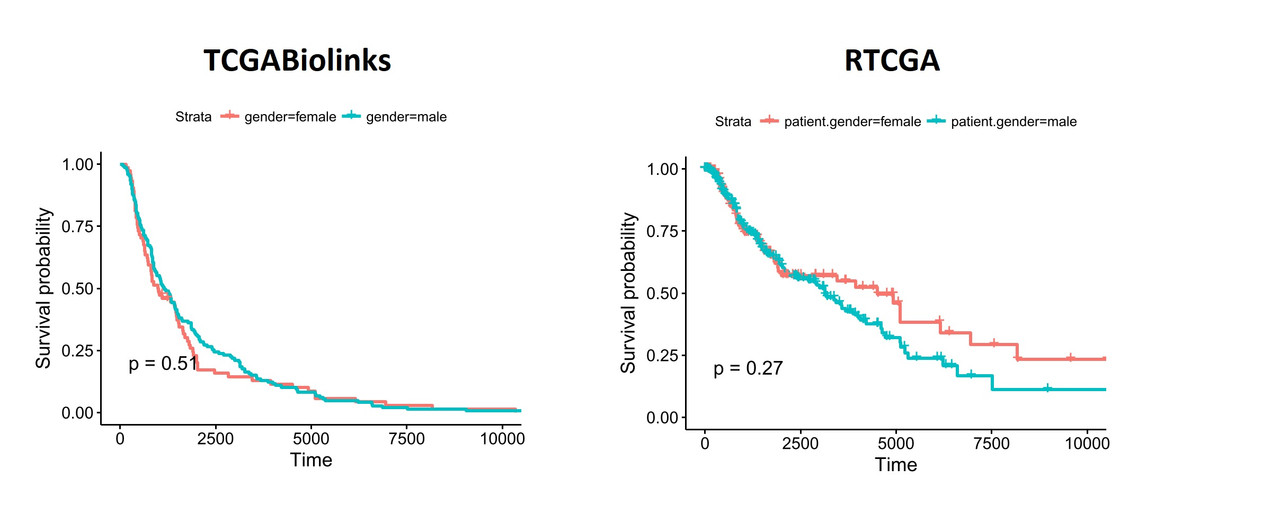
You can see similar differences when the data is segregated based on patient.gender:
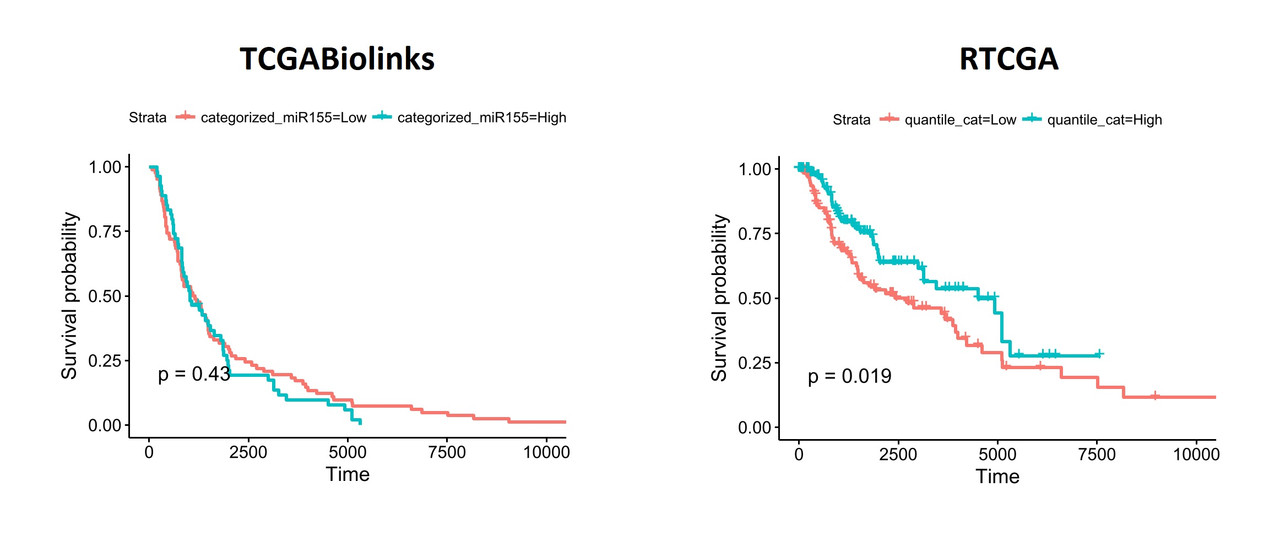
When I segregate patients based on the expression of hsa-mir-155 (top and bottom thirds), the differences become more obvious:
To understand what might be different in the datasets I exported the data from both packages and performed a comparison. Linked excel file shows the comparisons including clinical details (days_to_death and days_to_last_follow_up) and gene expression values of hsa-mir-155 (both read_count and reads_per_million_miRNA. I noticed that there are considerable differences between two datasets. I'm pretty sure, the way I organized the data is ok and I don't think the differences are due a mistake in data manipulation in R.
The code I used for analyses can be found at RTCGA_v1 and TCGABiolinks_v1
Please let me know why you think there is a discrepancy here. I'm pretty new to this type of analyses and hopefully, I didn't miss something silly.
Thank you very much in advance for your insights,
Atakan
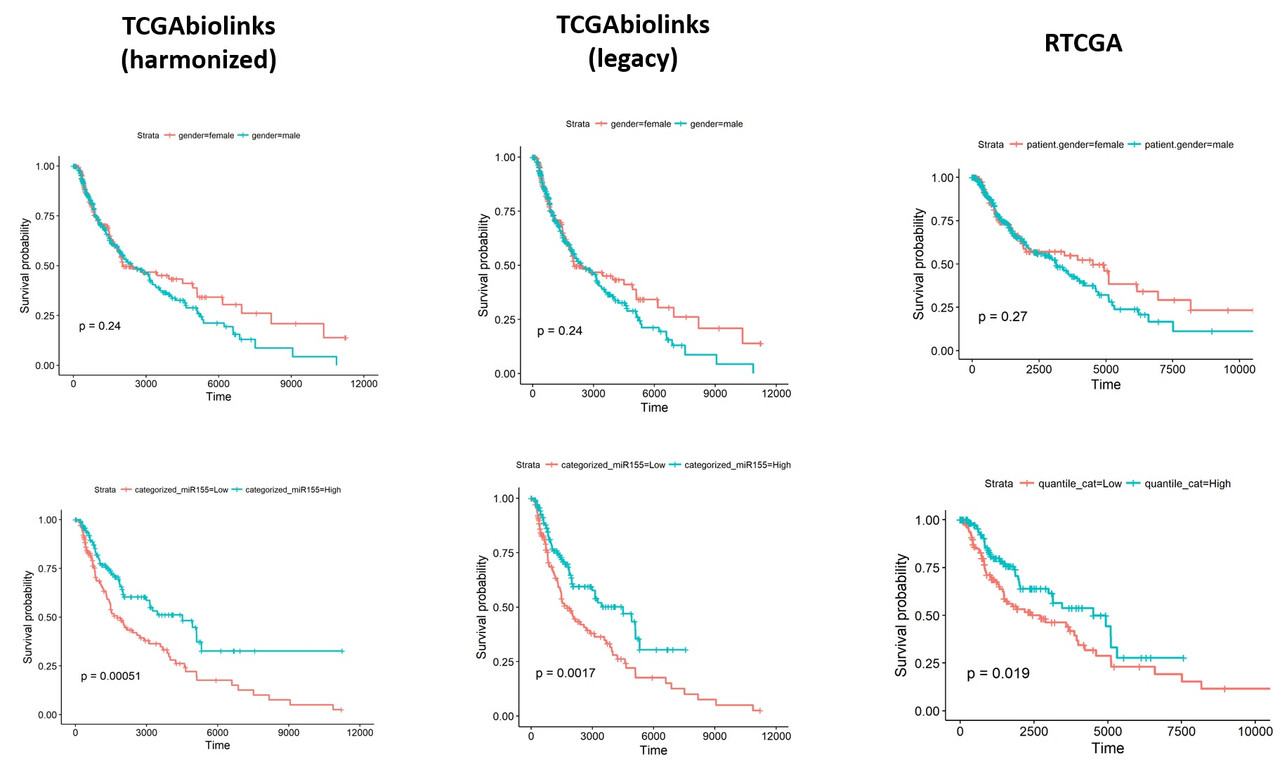


Thank you very much for the update
Dear atakanekiz,
it is nice to hear that you processed your data and find possible solutions on this matter-just an additional comment, in case you haven't used or tried the function TCGAanalyze_SurvivalKM- i think that includes anything you searched, as also the TCGAanalyze_survival -
Best,
Efstathios
Hello guys. As you are both using TCGAbiolinks, perhaps you could assist here: TCGAbiolinks TCGA-BRCA RNA-seq clinical data ?
I only used TCGAbiolinks once, a few years ago.