
I have been using DiffBind for differential-accessibility analysis with ATAC data and encountered the seemingly infamous normalization issue: our results are very different when normalizing by full library read depth as opposed to depth of reads within consensus peaks (from my understanding of how bFullLibrarySize param setting affects count normalization).
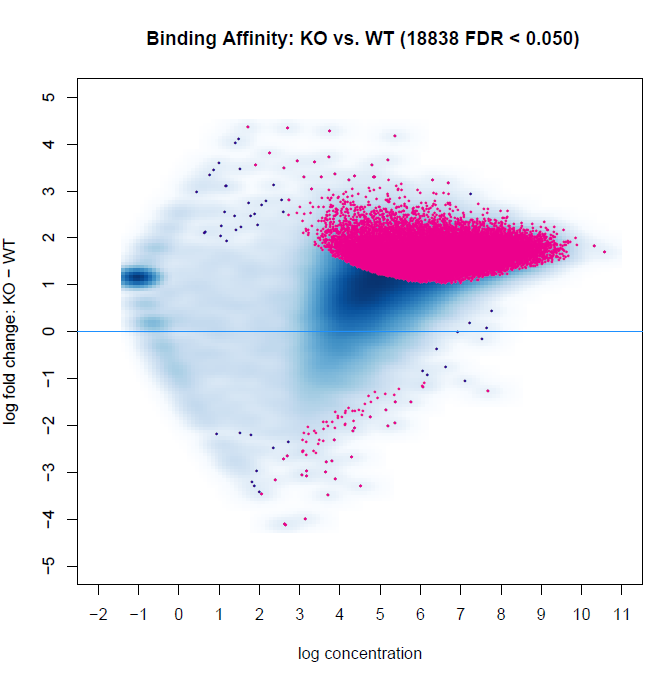
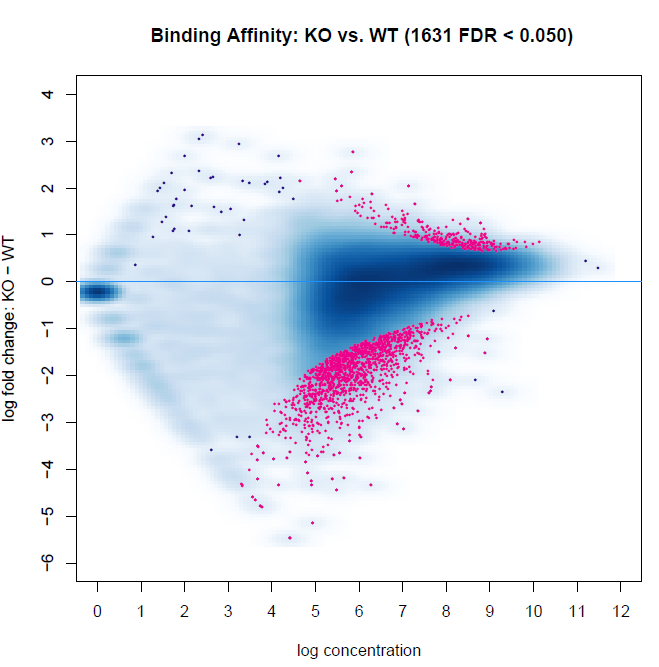
Below are two MA plots from bFullLibrarySize=TRUE and =FALSE using DESeq2, and we can see that the results are quite different. It would appear the background density normalization suggests to utilize the =FALSE method. We do observe variability of signal-to-noise between samples (FRiP ranges from 0.08 to 0.30 between samples). However, this may be indicative of the biology between experimental groups. This variability is also a reason why we have not relied on edgeR calculations, as I recall reading that Rory stated it is not an appropriate method for high inter-sample signal-to-noise variability within experiments. Does anyone have suggestions for interpreting these results, or which to favor?
I have a number of other flow cells which we also see variability from the outputs of bFullLibrarySize=TRUE vs. FALSE, so we are seeking insight for which to implement in scenarios with varying signal-to-noise intensities. I'm also planning to try csaw in the near future, so I can compare results to that method as well.
bFullLibrarySize=TRUE
bFullibrarySize=FALSE

I recently started using DiffBind for differential accessibility analysis of ATAC-seq. Could you elaborate more on why diffBind options are not robust and in which aspects csaw is better? I am still a newbie to this :)
Just out of interest, refreshing an old thread, when seeing this MA-plot from above (the 2nd, normalized one), would you say this (what looks like a) trended bias starting at logCPM ~ 6 is worth worrying about, e.g. to be corrected with loess as suggested in csaw?
I saw some of these trended bias in some datasets recently, still not sure whether I should bother with it or rather stick with default TMM if the bias is not too strong (means creating large FCs in the high logCPM range). Given this bias above was not biological it would probably make the regions on the topright (FC > 0, with large logCPM therefore high power) significant, while probably being a technical artifact (if we assume this is technical and not biological). Any thoughts?
I don't expect correcting this to cause too much of a change in the results, but it'd be worth having a look and testing that empirically.