
I have been trying to downsample my ATAC-seq libraries to normalize across all samples within my experimental design using ATACseqQC::readsDupFreq|estimateLibComplexity, which is based on the established preseqR::ds.mincount.bootstrap framework. After scaling all libraries to the same predicted complexity, I am now encountering some inconsistencies in the library statistics.
Basic experimental design is two experimental groups (treat vs. control) with two biological replicates in each group = 4 libraries.
My basic workflow is to trim and align (PE75, Bowtie2), then:
samples=("treat1 treat2 control1 control2")
for i in $samples; do
# sort by coordinates
samtools sort -T /tmp/${i}.sorted -o ${i}.sorted.bam ${i}.bam;
# remove mtDNA reads via Harvard ATACseq tool
samtools view -h ${i}.sorted.bam | \
python removeChrom.py - - chrM | samtools view -b - > ${i}.noMT.bam;
# sort by coordinates
samtools sort -T /tmp/${i}.sorted.noMT -o ${i}.sorted.noMT.bam ${i}.noMT.bam;
# index bam after mtDNA removal
samtools index ${i}.sorted.noMT.bam;
done
Then I assess for library complexity with the *.sorted.noMT.bam files. Here is a summarized output for total library and approximate common denominator within the experiment:
# full library
sample relative.size values reads
treat1 1 16233349 19698535
treat2 1 20637534 21479224
control1 1 20355060 22131151
control2 1 15925428 17488379
# scaled to normalize (predicted)
sample relative.size values reads
treat1 0.98 15943266 19304564
treat2 0.77 15970045 16539002
control1 0.78 15981835 17262297
control2 1 15925428 17488379
... where relative.size is the relative library size that yields specified values (estimated library size) and reads (estimated reads to achieve that library size).
I then subsample the files via samtools view -h -b -s:
samtools view -h -b -s 0.98 treat1.sorted.noMT.bam > treat1.sub.sorted.noMT.bam
samtools view -h -b -s 0.77 treat2.sorted.noMT.bam > treat2.sub.sorted.noMT.bam
samtools view -h -b -s 0.78 control1.sorted.noMT.bam > control1.sub.sorted.noMT.bam
# leave sample control2 as is (lowest complexity sample)
... and reassess complexity on subsampled files:
sample relative.size values reads
treat1.sub 1 15944465 19304847
treat2.sub 1 15962582 16540771
control1.sub 1 15985716 17264865
control2 1 15925428 17488379
Looks like it worked. Excellent!
I then remove PCR duplicates via Picard MarkDuplicates, name-sort reads, samtools fixmate to facilitate downstream conversion to BEDPE for MACS2 peak calling:
samples=("treat1.sub treat2.sub control1.sub control2")
for i in $samples; do
# remove duplicates
java -jar picard.jar MarkDuplicates \
I=${i}.sorted.noMT.bam \
O=${i}.noDups.noMT.bam \
M=${i}_dups.txt \
REMOVE_DUPLICATES=true;
# name-sort reads
samtools sort -n ${i}.noDups.noMT.bam ${i}.sorted.noDups.noMT;
# fix mates
samtools fixmate ${i}.sorted.noDups.noMT.bam ${i}.fixed.bam;
done
Lastly, one final coordinate sort and index of the BAMs for csaw:
for i in $samples; do
samtools sort -T /tmp/${i}.sorted.fixed -o ${i}.sorted.fixed.bam ${i}.fixed.bam;
samtools index ${i}.sorted.fixed.bam;
done
Later on, a discrepancy is observed when we assess library size in csaw:
sample lib.size
treat1.sub 17203692
treat1.sub 16057876
control1.sub 19706273
control2 19832246
... which is a summarized output of counts$totals.
Does this indicate we did not successfully compensate for library complexities by subsampling? Why are the calculated inter-sample library sizes different after subsampling?
Using samtools flagstat, I have tried to investigate this further. Here is a breakdown for sample treat2:
*.bam *.noMT.bam *.sub.noMT.bam *.sub.noDups.noMT.bam *.sub.fixed.bam
82159808 82159808 63269230 59839102 59839102 total
0 0 0 0 0 secondary
0 0 0 0 0 supplementary
0 0 0 0 0 duplicates
80032898 70040719 53936734 50506606 50506606 mapped
82159808 82159808 63269230 59839102 59719666 paired in sequencing
41079904 41079904 31634615 29905399 29859833 read1
41079904 41079904 31634615 29933703 29859833 read2
79045966 69160532 53259798 49963952 49963952 properly paired
79362718 69417198 53457164 50146472 50146472 with itself and mate mapped
670180 623521 479570 360134 360134 singletons
82172 72510 55718 53420 53420 mate mapped diff chr
27719 27056 20902 20302 20302 mate mapped diff chr (mapQ>=5)
... or partially presented as a graph:
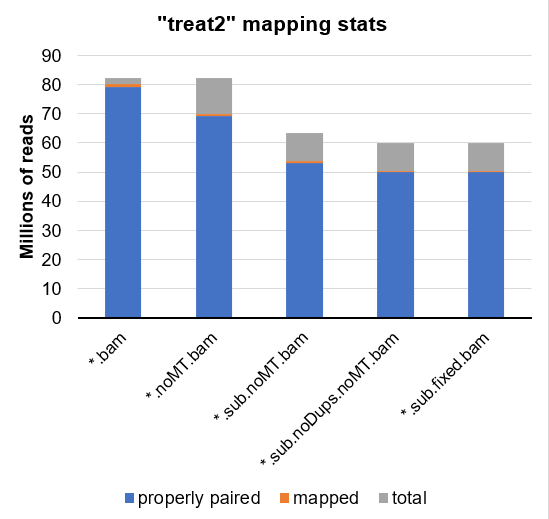
Could it be that the mtDNA are being read into csaw as true reads, even though they are not labeled as mapped?
Here is another look at all the final BAMs in this experiment which are subsequently loaded into csaw:
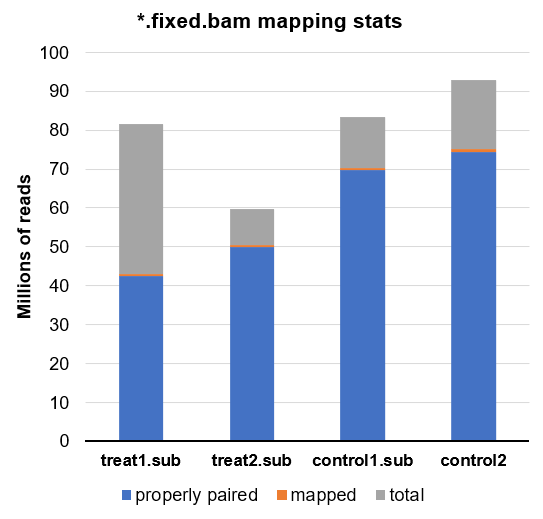
Comparing this to the library sizes calculated by csaw, we can tell even visually that the calculated sizes do not appear to match the read depth with or without mtDNA and whether or not it is utilized.
sample lib.size
treat1.sub 17203692
treat1.sub 16057876
control1.sub 19706273
control2 19832246
# these are the same calculations from above
In conclusion, I do not understand why these libraries are showing highly variable read depths and library sizes when they should have been theoretically normalized to complexity. I am seeking explanation for this phenomenon and ultimately if I should trust 1) calculated library complexity estimates from preseqR/ATACseqQC or 2) csaw library size calculations. In the case of the latter, how can I correct my method?