Entering edit mode

5.8 years ago
marina.v.yurieva
▴
570
I'm trying to put genes from ENCODE and NONCODE in one gtf file and filter out NONCODE genes which are already in Encode. My bedtools command is:
bedtools intersect -wa -a NONCODE.gtf -b Gencode_exon.gtf -v -s -f 0.9
(I only take "exon" features from Gencode, otherwise it removes more transcripts from NONCODE)
This works okay for the most part but if I have overlapping transcripts in my NONCODE file, I get only one exon in the output:
Does anybody have any idea why does it happen and how to fix it? I can use another tool, just need to make sure I get all the Gencode genes and only the transcripts from NONCODE which don't completely (90%) overlap with Gencode. It has to be strand-specific too.
If you want the GENCODE transcript where there's overlap, then why are you using
-wawith-abeing your NONCODE list?-aand-bshould be flipped, no?I also question the use of
-v...To get what you want, I think that you need
-wao, with-a GENCODEand-b NONCODEThank you, Kevin! I tried that but it doesn't really work the way I need: I guess, when bedtools works with gtf file, it doesn't really pay attention to "gene", "transcript", "exon" fields and doesn't see the relations between them... If I use Encode file with "gene" and "transcript" fields, it overlaps it with the whole thing, not paying attention that exons are not along the whole gene and have breaks. If I use only "exon" fields, then I'll have to write another script of calculating the overlap of all the exons in the gene and, if they overlap > 90% exclude the whole gene_ID from the original file. I was wondering if there is already a tool which does all that...
Some example data and expected output would help to understand and resolve the issue better @OP
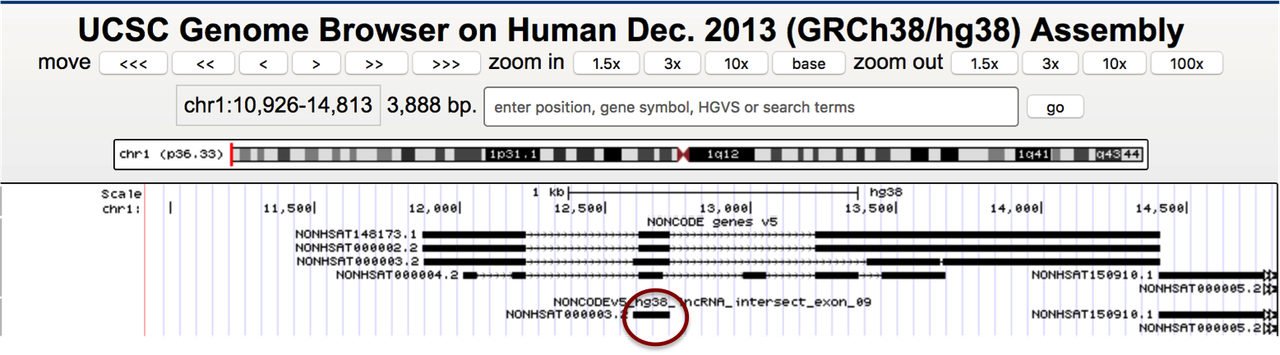
Sorry, I'll try to explain again. I need to add the transcripts from NONCODE to Gencode database but have to make sure the ones which overlap with Gencode > 90% are not included (the transcripts in the circle should be excluded). The closest I've gotten to this by using
But that option excludes some original exons from the NONCODE transcripts (see exons with the arrows), and I need to add the original full transcripts, not modified.
I tried to use the suggested option
bedtools intersect -wao -a Gencode.gtf -b NONCODE.gtf -sbut it gives an overlap of all the features with everything (genes with exons, transcripts with exons, etc) and will require a lot of downstream parsing. I don't really know if any other software can do an overlap looking at the exons from one transcript but bedtools doesn't seem to really do that.