I have stranded RNA-seq data which doesn't look stranded when I visualize bedGraphs in Genome Browser:
I tried different aligners (star, bowtie, bwa), different ways to make bedGraph files, nothing works. But when I run salmon on these samples, it detects ISR (paired-end stranded library). I'm very confused why it doesn't look stranded in the browser but I know is stranded. Any help will be really appreciated! I've been trying to solve it on my own for a while...
The reason for this is that whether or not to consider a fragment for quantification in salmon is considered after the mapping is written. That is, the reads are mapped to the transcriptome, without regard for the inferred or provided library type, and then these mappings are written to file if requested by the user. However, when salmon considers the probability of these mappings arising from different transcripts, it will discard those that are not compatible with the library type. So, my guess is that you're seeing in this output multi-mapping reads where the sequence matches, and hence there is a possible allocation of this read to the transcript, but for which these mappings are later discarded during quantification. You could, for example, filter the mapping file based on the tags of the alignment records, to only keep those reads that align according to the ISR library type.
Well, you should definitely filter on more specific tags to get rid of the alignments that don't map according to the library type, since salmon will discard them for quantification purposes. It's not easy to discard the multimappers. The fact that a multi-mapping reads maps to a particular locus doesn't mean that it was assigned there for the purposes of quantification. To determine that, you'd have to weight each multi-mapping read according to the abundances estimated by salmon. However, simply getting rid of the reads that align different from the library type should remove some of the ambiguity about how the pile-ups should look.


This is quiet uninformative. If "nothing works", then please provide the codes you used. How did you create the bedGraphs (exact command please)?
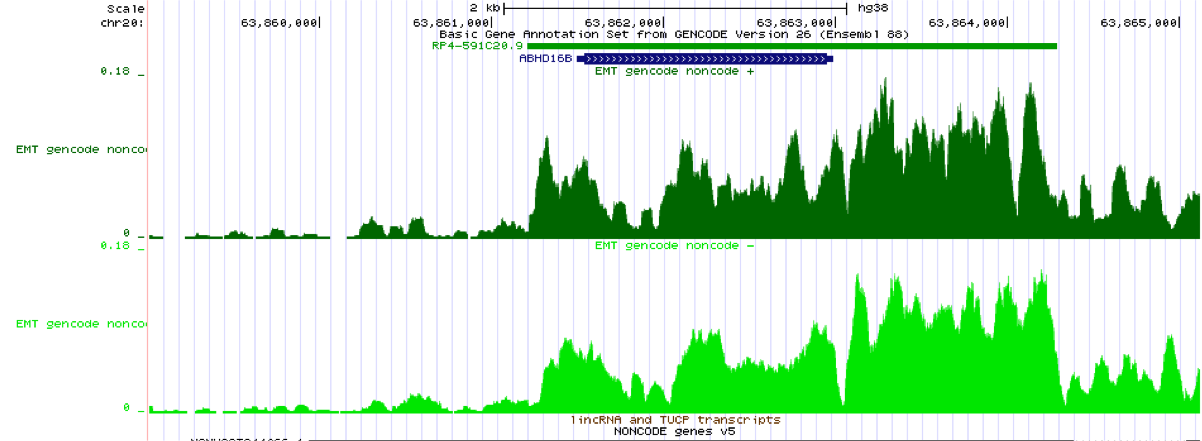
You should check a known housekeeping gene like ACTB or GAPDH before you check some obscure transcript that may not be real.
Yeah, I looked at everything, just gave the first example. Here is Gapdh:
And how are you generating these bedGraphs?