Entering edit mode

5.5 years ago
marongiu.luigi
▴
710
Dear all,
I have extracted reads that did not mapped to a reference genome with
samtools view -h -f 4 <aln-srt>.bam <unmap-srt>.bam
where the input file is sorted. Then I created the fastq files with:
bamToFastq -i <unmap-srt>.bam -fq <Ump_1>.fq -fq2 <Ump_2>.fq
gzip <Ump_1>.fq
gzip <Ump_2>.fq
This created many warning errors such as:
*****WARNING: Query HWI-ST1437:64:C3UM1ACXX:4:2315:4948:54750 is marked as paired, but its mate does not occur next to it in your BAM file. Skipping.
but overall, the procedure was completed and I could re-align to another reference with:
bwa mem -t 8 -R $RG <ref.fa> <Ump_1>.fq <Ump_2>.fq | \
samtools sort -o <newaln-srt>.bam
and I got alignments such as this (from IGV):
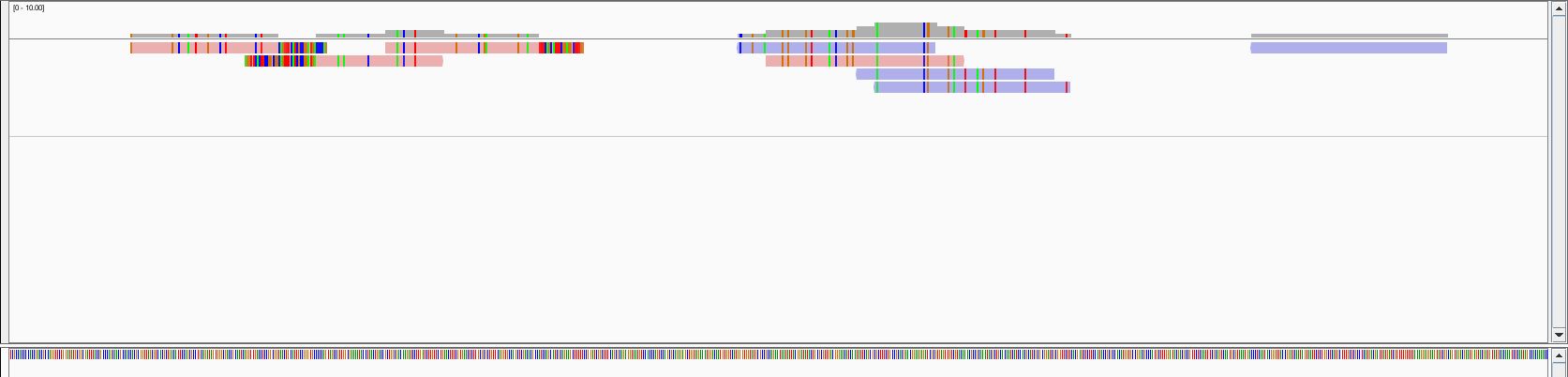
So far so good, i'd say (unless there is a flaw in this process of course).
I then tried to apply a recalibration with the following steps:
sambamba markdup -r --overflow-list-size 1000000 --hash-table-size 1000000 \
<newaln-srt>.bam <newaln-srt-dedup>.bam
gatk BaseRecalibrator \
-R <ref>.fa \
-I <newaln-srt-dedup>.bam \
-O <newaln-srt-dedup>.tab \
--known-sites <ref>.vcf.gz
But I got:
[...]
Running:
[...]
.BaseRecalibrator done. Elapsed time: 0.51 minutes.
Runtime.totalMemory()=1161822208
***********************************************************************
A USER ERROR has occurred: Input files reference and reads have incompatible contigs: No overlapping contigs found.
reference contigs = [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, X, Y, MT, CHR_HSCHR15_4_CTG8, CHR_HSCHR6_MHC_SSTO_CTG1, CHR_HSCHR6_MHC_MCF_CTG1, [...]
423.1, KI270392.1, KI270394.1]
reads contigs = [V]
***********************************************************************
Set the system property GATK_STACKTRACE_ON_USER_EXCEPTION (--java-options '-DGATK_STACKTRACE_ON_USER_EXCEPTION=true') to print the stack trace.
So the question is: how can I deal with this error? Shall I do the recalibration in the first place?
Thank you.

Good spot, it must be something with the headers in the reference. I'll try again. Thank you