Both Genrich and MACS2 can call peaks on paired-end ATAC-seq data and remove duplicates. Multi-mapped reads are typically removed/disregarded during the alignment step (i.e. bowtie2). Removing mitochondrial reads can be accomplished with multiple methods of which I prefer using sed but I can see how having this wrapped into the peak caller is convenient. I have tried both methods with ATAC-seq data.
An important difference is that MACS2 will call peaks on the paired end fragments and Genrich will call peaks on the cut sites (5' end extensions, set with -d, of the paired end fragments). With MACS2 you can use some suggested options to recapitulate the Genrich approach and calling peaks with Genrich 'not' in ATAC mode may give you similar MACS2 results as above. Overall each method above on the same bam file yields different peaks (as one would expect)
Downstream we supplied the peaks files to DAStk and were more satisfied with the MACS2 results but a bit unnerved by the differences.
Although our downstream differential transcription factor (TF) analysis results remained similar between both peak callers we found that MACS2 called more peaks and we validated these peaks from ChIPseq data of TFs. We reasoned that if an ATAC-seq peak overlaps a ChIP-seq TF peak then the peak caller is providing its intended use from the BAMs. Also, an increased number of peaks detected probably happens in part because one long Genrich peak sometimes overlaps multiple smaller MACS2 peaks (especially at gene promoters). I only theorize but cut site enriched peak calling may in effect "flatten" the peak summits that are called by MACS2 (with parameters like -f BAMPE and NOT --shift -100 --extsize 200 for cut site enrichment).
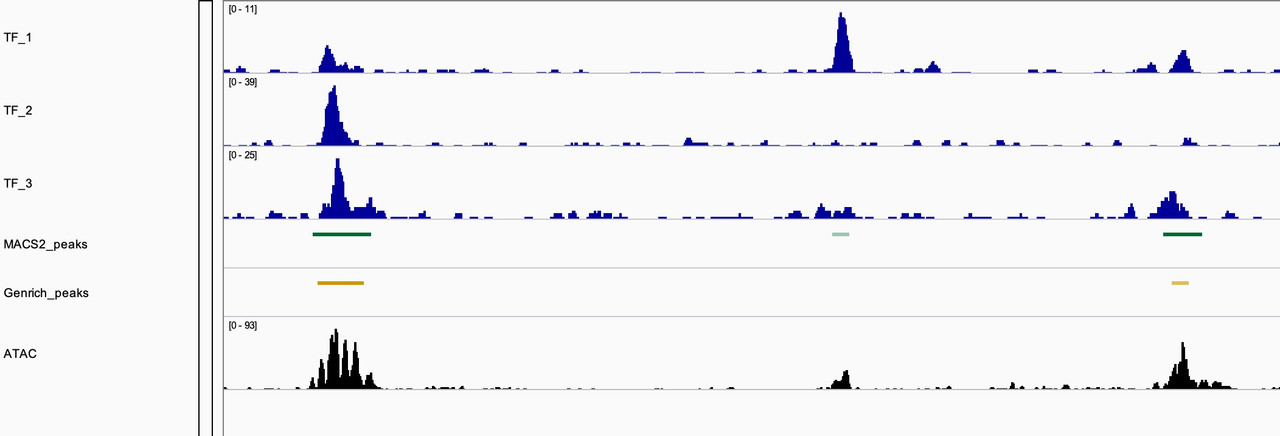
They certainly on average tend to overlap with MACS2 peak caller. But we found sites where Genrich did not call peaks when we clearly have ChIP-seq data of TFs overlapping. As seen in the image, the middle shows that Genrich did not call a peak where MACS2 did, and there is evidence of a TF bound at that site and an appreciable ATAC-seq peak.
1) I have ATAC in duplicate from multiple cell lines.
2) We find MAC2 called more peaks than Genrich and these peaks were validated with ChIP-seq data of TFs. Consistently MAC2 yielded 100K peaks and Genrich 50K (per50mil reads, although peaks platuea genome biology).
One would really need a proper benchmark paper addressing this. macs calls a lot of "peaks" that visually look small on the browser which I always found questionable. A study checking the biological meaning of these would help a lot, hope someone addresses this soon. Still, I would probably rather have fewer but higher confidence peaks as peak numbers are in any case, as you say, high, in the tens-of-thousands range.
As for this TF overlap, a TF binding site does not necessarily imply open chromatin. There are well-known factors that bind closed chromatin so I doubt TF overlap is a good proxy to validate ATAC-seq. It might even make sense that TF binding somewhat poises the region to open up upon a stimulus (maybe attracting some few factors but not the entire chromatin remoddeling machinery), but in its absence of that stimulus the region is borderline-accessable so that fuzzy/noisy ATAC-seq signals might be expected (questionable if this then qualifies as a true peak).
So what do both individual ATAC-seq replicates look like for the snapshot you showed above? I agree with ATpoints notion that MACS tends to be on the lenient side, which is probably aggravated by the fact that it does not integrate information from replicates.
I would generally agree that TF binding more often than not indicates open chromatin (there are relatively few pioneering TFs), but the whole issue of biologically meaningful sites of open chromatin is a whole different issue. I guess the take-home message for now is that Genrich tends to be a bit more conservative.
The duplicate data is consistent with both peak callers. I agree that the complexities of ATAC data can be independent of TFs and that some TFs may provide open chromatin states while others may not. ATAC and ChIP data would be interesting to analyze alongside depletion conditions. I think moving forward I'm more curious about the difference in peak calling method. The snapshot above is of MACS2 peaks called with -f BAMPE setting while the Genrich peaks are inherently cut-site enriched. I think analysis of biological significance as well as comparison of these two methods would appreciated.
while the Genrich peaks are inherently cut-site enriched
True, as it counts cutting sites and then extend these by (default) 100bp to smoothen the signal. Similar behaviour can be achieved with macs by extracting the cutting sites from the BAM (so 5' end of each read), saving them as BED and then use -f BED, forcing macs to indeed count cutting sites, followed by smoothing with shift/extsize. Still, I think the elegant thing with Genrich is the abiity to handle replicates and by this increase confidence in peaks without external tools such as IDR (which can only handle n=2).
I just learnt of Genrich this week - we installed it (which went smoothly), ran it and got results that didn't seem off. So, generally, I would recommend it although you may not want to use it for the super-high impact paper that you are submitting next week because it does not seem to be published yet and reviewers may complain about that.
I should add that the reason we tried it out was that we found these arguments to be convincing and hitting all the complaints we've had about MACS for PE data, especially in the ATAC-seq context.
Thank's for the Genrich. I am not after any specific functionality, in general I have found that MACS2 doesn't work quite well with H3K9me3 or H3K27me3 marks (might be more). I don't know what the issue is and how it can be fixed. On the other hand PeakRanger (CCAT) works quite well with these marks.
H3K9me3 and K27me3 yield very broad and much more subtle enrichments than, say, K4me3 or ATAC-seq for that matter. It's known that MACS is not the best performer for these broad marks, but the narrow and strong enrichments that ATAC-seq should yield are typically handled well by it.



For paired end reads, specially ATAC-seq, you can use fseq: http://fureylab.web.unc.edu/software/fseq/
How is
F-Seqspecific for paired-end data? Doesn't it use BED files as input requiring additional preprocessing steps?