
Hello! I'm quite new to NGS data analysis and have a few questions about certain modules in FASTQC.
I want to know the importance of analyzing the per base sequence content, which measures the percentages of A, T, G and C throughout each base in the read and also why we check the per sequence GC content. I read the manual provided at the FASTQC website but I would like to understand more about why they are done.
From the manual, I get that the A,T,G and C %s shouldn't vary much throughout the bases for a good sequencing project. Too much variation between the Watson-Crick bp compositions means that there is some kind of bias in the library (the manual goes on to list a few). Is there any other reason why this is done?
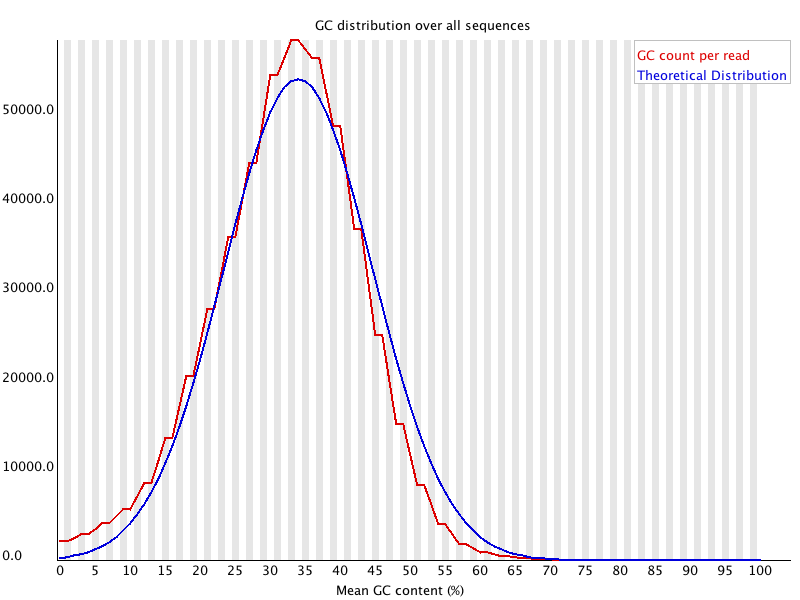
As for the GC content, I would like to know why we particularly look at GC% again since we already looked at the composition in the previous per base sequence content module. The manual here says that the GC% of the data should be a normal distribution. Is this module one way of checking for GC bias that occurs during Illumina sequencing? Can we also conclude that the sequencing quality is good when the curve obtained for our data matches the GC% of the genome when known? Is there any other reason we do this? Also, in the manual it is mentioned that:
A warning is raised if the sum of the deviations from the normal distribution represents more than 15% of the reads.
Here by 'sum of the deviations', they mean the standard deviation right? Or do they mean the deviation of the GC content per read from the theoretical distribution?
Thanks in advance!
The only thing I look at in
fastqcare the adapter content and overrepresented sequences plus the per-base quality scores to see if there is any systematic issue with the run or at a certain cycles. All other metrics do not really add information (at least for me). Best is to proceed with your downstream analysis and see if that works well.Wow that's news to me. Thanks!