
Hi all.
Recently, I got sequence for H3K27ac ChIP-seq data performed in various stimulation and knockdown condition. I have processed the reads and did peak calling using macs2. Looking into IGV browser, the enrichment looks good and the peaks are identified. The peaks called are of in the size range from ~1kb to 10kb. My concern here is whether the size of peaks identifies is really broad or is it ok to get large size peaks from H3k27ac chip. Next if want to perform motif identification, should I take all the broad peaks or perform peak calling to get broad peaks of lesser size (~1kb).
I would highly appreciate your suggestions and feedbacks
Thanks
peak calling method:
ls bowtie_out/*.25M.bam | cut -d'/' -f2 | parallel --verbose 'macs2 callpeak -t bowtie_out/{} -c ../Input_R1.bam -g mm -n {=s/.25M.bam//g=} --verbose 2 --broad --broad-cutoff 0.01 --cutoff-analysis --fe-cutoff 4 --outdir macs_peak'
IGV browser snapshot:

Zoom of portion highlighted in above image:

free image hosting

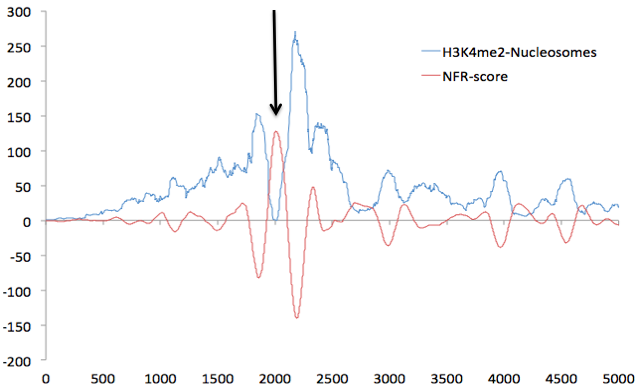

The motifs bound by TFs are most likely within the valleys of your H3K27ac signal. This hypothesis has been tested (article) and a program called EpiSAFARI was developed to identify peaks within valleys . You can certainly do motif analysis on the peak set you have but it wouldn't make must biological sense. I also want to point out that this is true for H3K27ac but may not be true for other histone modifications.