Entering edit mode

4.5 years ago
marongiu.luigi
▴
710
Hello,
I need to extract reads from an alignment I have done of the whole human genome. I am looking for the gene MACC1 which is located at Chr 7 20134655..20217384, I used this command:
samtools view -b -h <AlnSrt.bam> "Chr7:20134655-20217384" > <subSet.bam>
but I got:
[main_samview] region "Chr7:20134655-20217384" specifies an unknown reference name. Continue anyway.
Using IGV I can see that the reference has a name chr7 so I changed to:
samtools view -b -h <AlnSrt.bam> "chr7:20134655-20217384" > <subSet.bam>
But in that case, nothing happens at all. The unknown reference name comes out even if I use 7:20134655-20217384.
How can I check the name of the reference?
How can I extract these reads?
Thank you
So I got
@SQ SN:chr7 LN:159345973since the length is 159 345 973 and I am looking for 20 134 655, it should work with samtools view -b -h <alnsrt.bam> "chr7:20134655-20217384" > <subset.bam>Is it possible there is simply nothing aligned against that portion of the genome?
Try expanding the range, eg 1Mb either side.
chr7:20034655-20317384
If still nothing, go further. Also do other regions you pick work? If nothing works then maybe the index is broken.
I am not getting anything even on the other chromosomes, but I can see the reads on IGV, which uses an index: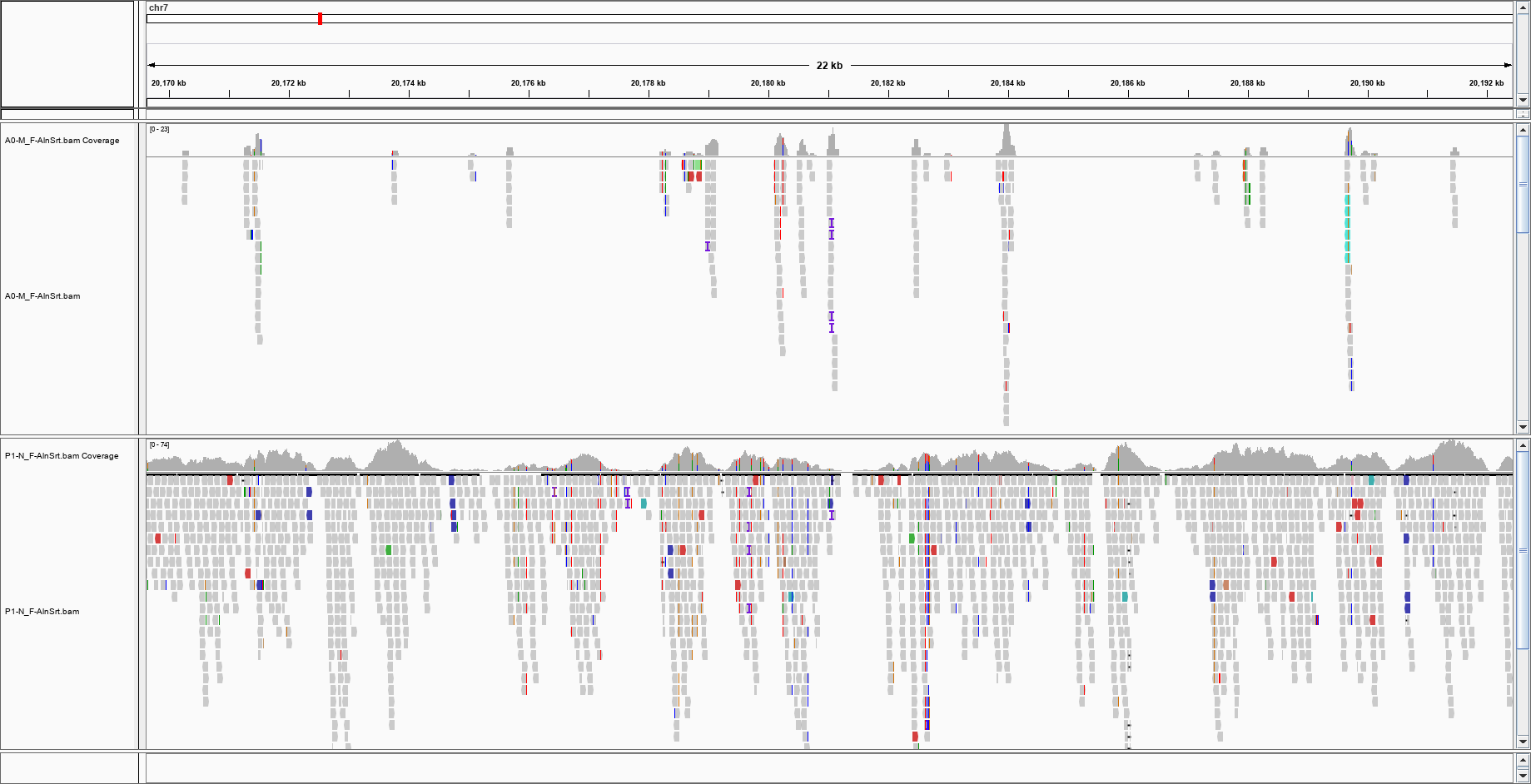 I can see that there are reads in this region; how can I check if the index is broken?
Anyway, even with the error, a file has been created:
I can see that there are reads in this region; how can I check if the index is broken?
Anyway, even with the error, a file has been created:
Would that be trustful?
Thank you
When you say that nothing happens, do you mean that nothing appears on the terminal ? It is actually what is expected when the command works (there is no error or warning). This is probably when the
subSet.bamfile has been created. To make sure that it comes from this command and not from something else, deletesubSet.bamand run the command again. By the way I don't think you need to quote (" ") the region insamtools view.Good point, I'll try that. Thank you -- Update: it worked.