Entering edit mode

4.4 years ago
robert.murphy
▴
80
What is the difference between paired end reads and overlapping reads, after reading around I am struggling to find an answer as to what overlapping reads actually are. From what I can see they just seem to be another term for paired end reads but I feel this must be wrong or I would have found somewhere stating this?
Secondly, why merge overlapping reads prior to assembly?
Any help would be appriciated :)

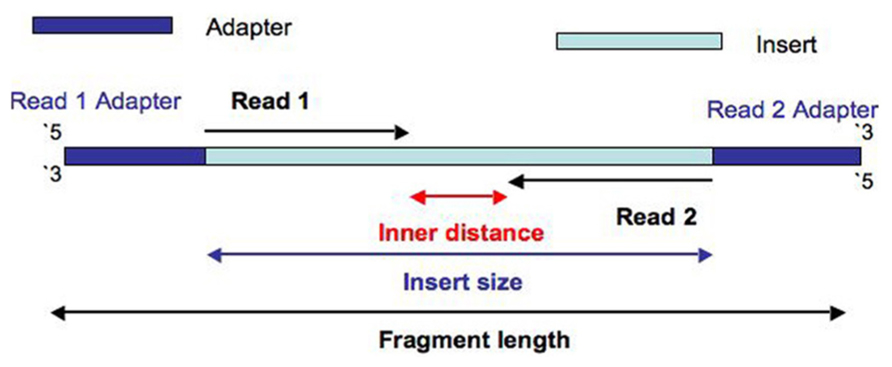
Paired-end reads - reads that are produced in pairs with approximately known insert distance (they are designed in this way). Overlapping reads - paired-end reads when the insert distance becomes negative (they physically overlap). Why to merge them - to avoid mis-assembly. Imagine a read that has ACGT sequence and has an overlapping read of TACG. You can merge it into ACGTACG. Or you can remain it the same - and then another read, e.g., CGTCC may occur in your data, and you will get an assembly path ACGTCC instead of correct ACGTACG. I don't say that's how the assembly algorithm makes a choice where to put a path (there are various considersations), but it may happen in approx this way.
UPD: instead of insert distance should be inner distance, as clarified by the answer below.