
Hi, we're working on a scRNA-Seq samples, where R1 has 16bases with the cellular and molecular barcodes, while R2 is 150bases long and contains the genomic sequence.
This data set appears to be very problematic, as it shows many problems. We think that we have sequence into the adapter, as running fastqc shows an over-representation of PolyA stretches and a drop of quality as one can see in the first attachment.
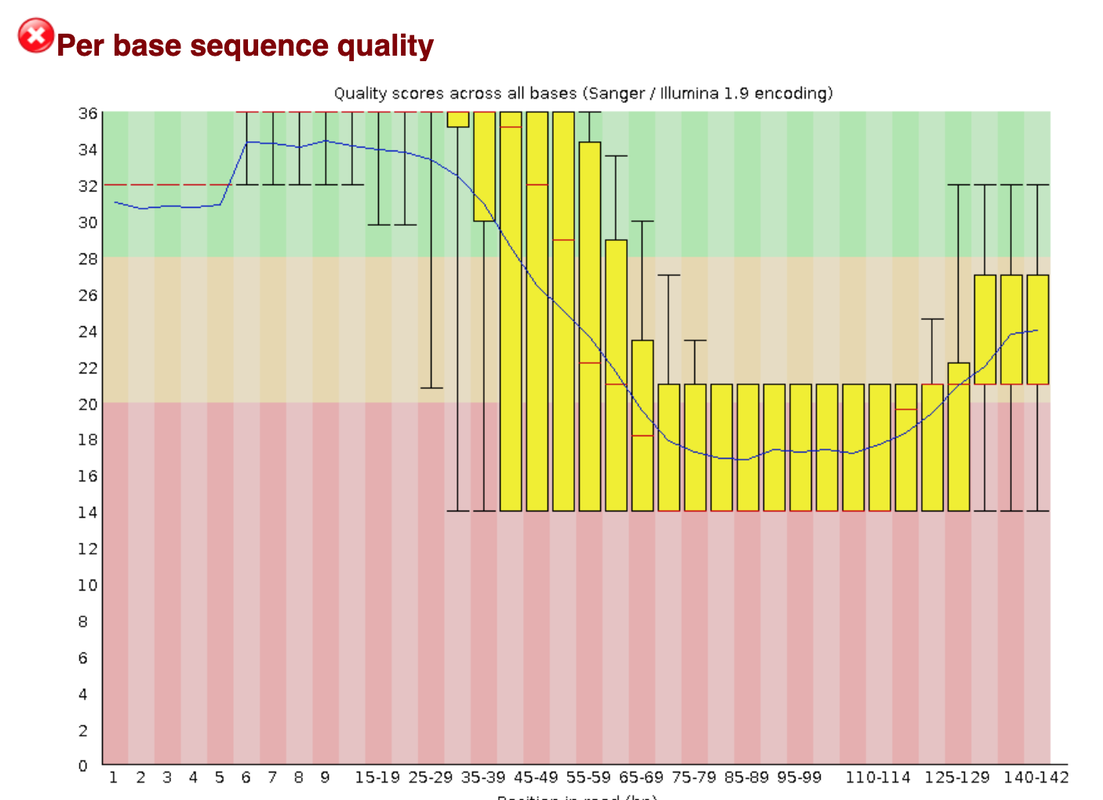
For that reason we would probably hard trim the samples (maybe after qc-trimming)before the quality drop. But what we don't understand is why our R1 shows only stretches of N.
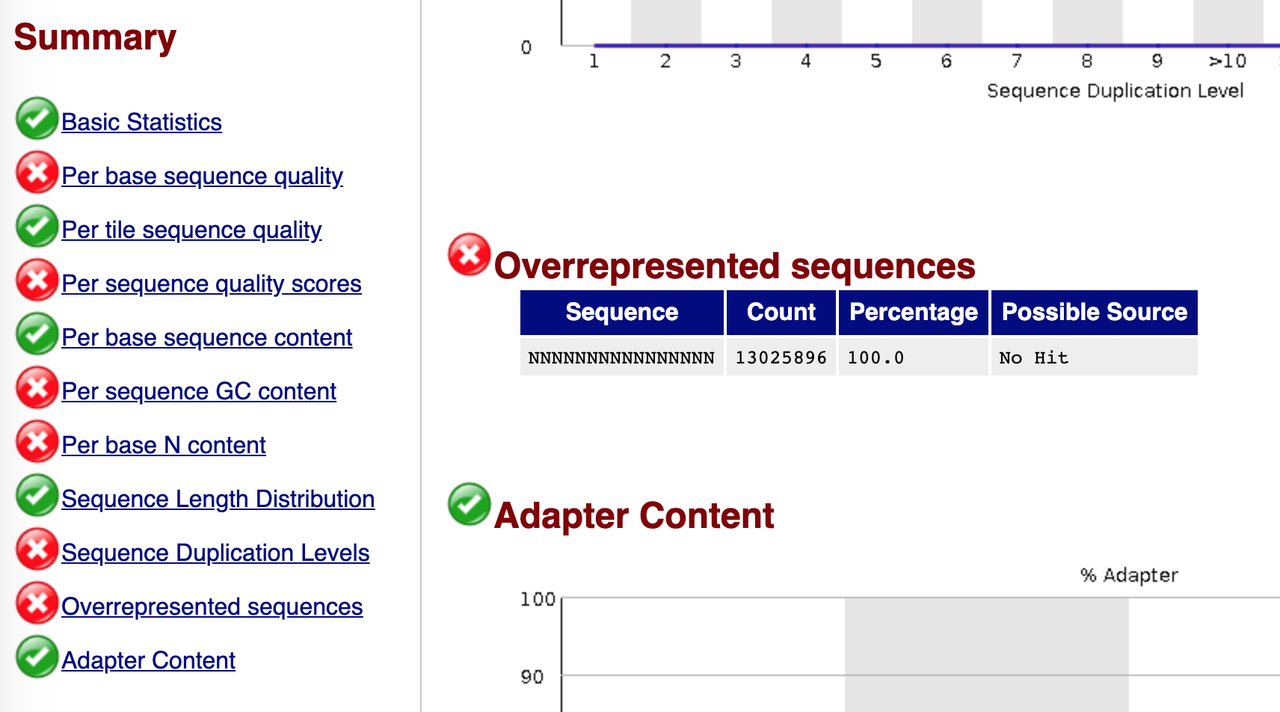
Does anyone has an explanation for this kind behavior? Has anyone seen something like this before? the bcl2fastq did not show any errors at all and the fastq is just a long list of read with ployN stretches.
thanks Assa

Do you have access to the library QC? (pre-sequencing)
yes, but the QC is good