Entering edit mode

4.1 years ago
tommaso.green
•
0
Hi,
I'm trying to make several pairwise global alignments using biopython Pairwise Aligner. I'd like to store my results in a phylip format so that I can use the output to build a phylogenetic tree using Phylip. The problem is that I can't seem to be able to write my alignments using AlignIO package, the write function probably doesn't want my PairwiseAlignment object. The error I get is "object of type 'PairwiseAlignment' has no len()". What can I do? Thank you for your time.
from Bio import SeqIO, AlignIO, Align
from itertools import combinations
cov_sequences = list(SeqIO.parse("gisaid_cov2020_sequences.fasta", "fasta"))
combos = list(combinations(cov_sequences, 2))
aligner = Align.PairwiseAligner()
alignments = aligner.align(combos[0][0].seq, combos[0][1].seq)
with open("alignments.phy", "w") as handle:
AlignIO.write(alignments, handle, "phylip")
print(alignments)
Hi, is there a reason you don't use multiple sequence alignment?
Is this similar to what you need? pairwise alignment for multiple sequences in a file
Yes indeed, but my problem is that I want to write all these alignments to a phylip file
The main reason is I'm doing this project for a course and the prof recommended using global alignment since I'm trying to align RNA sequences of the same species (coronavirus).
Cool. What does the output of the alignment look like if you print it? It would be helpful to print sequences (e.g. combos[0][0].seq) and alignment before writing into the file to make sure they are correct.
Thank you Fatima for your kind attention. I just started a bioinformatics course and I didn't expect this much help and interest.
You'll find attached a screenshot of the outputs you asked + the output of alignments[0] (truncated for obvious reasons).
I have some doubts about what I should really do. The prof asked to produce a phylogenetic tree (using Phylip if possible) using pairwise global alignments of the virus sequences. My question is: do I have to produce a .phy file for each pair or is it possible to store all pairwise global alignments in one phylip file? Thank you very much.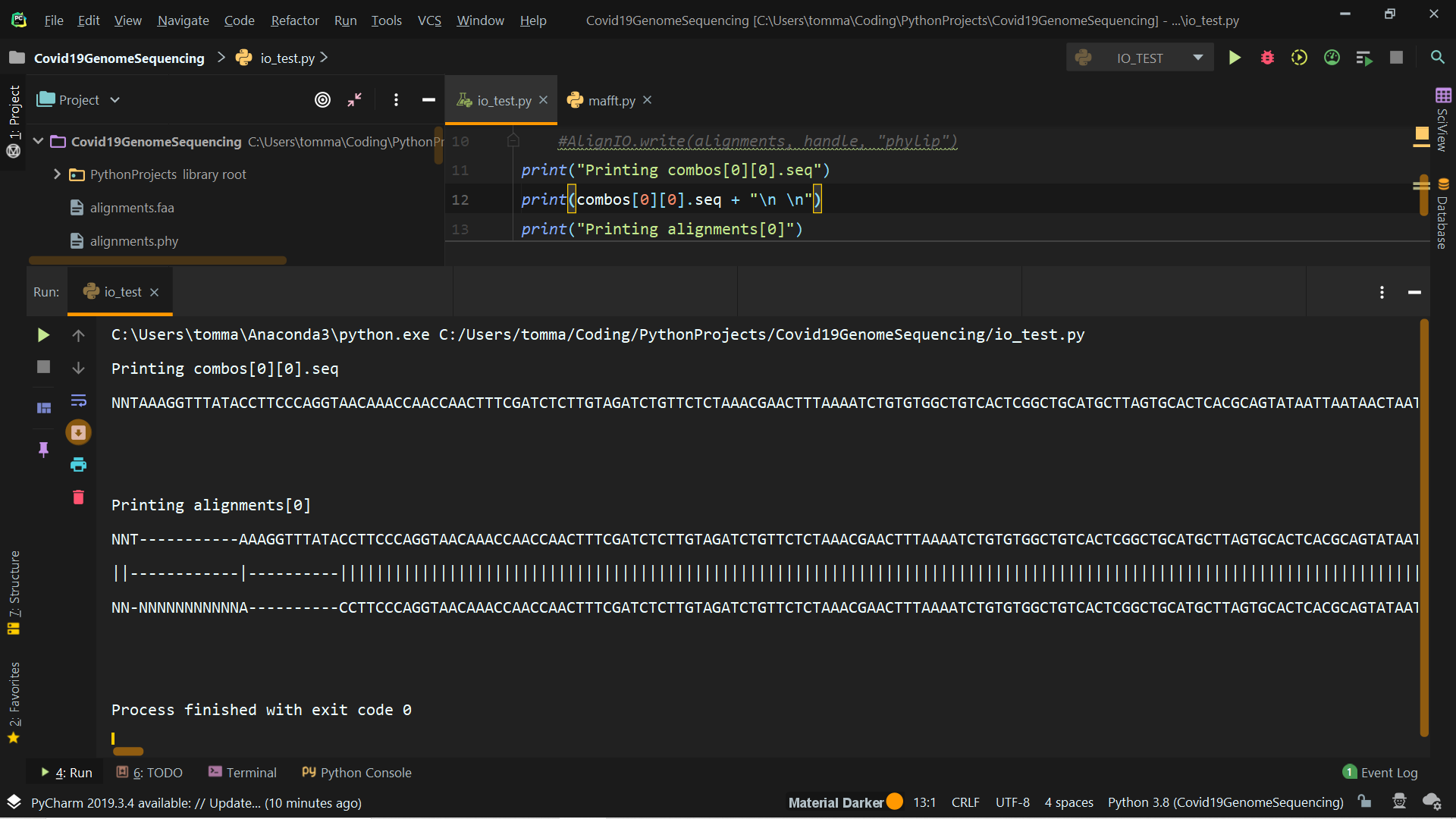
YW :)
I'm not sure, but I think the problem is that you're using the script that gives you the alignment, which is good if you only want to see the alignment or count the number of gaps, .... but if you want to save this alignment into a file in a recognizable format and not just raw text, you also need to have a header for your sequence to be able to save it in fasta or phylip format.
You might be able to use this function but I'm not sure if it's compatible with your alignment:
or you can read them from a file, for example:
http://biopython.org/DIST/docs/tutorial/Tutorial.pdf
6.5 Pairwise sequence alignment
I know the following question is different from what you're trying to do, but you can use it to get some ideas:
Text file to Phylip format
I know about the header, the thing is that AlignIO.write() function takes care of that too but it only works for multiple sequence alignments not pairwise. Anyways I'm probably abandoning Python for this project (see other answer below).
Do you have to align sequences individually and do you have to use biopython for this? You'd never need to do this 'in the wild', if its an assignment, its a slightly silly one...
Actually I am guessing from the assignment that what needs to be done is to do pairwise alignments of all the sequences, generate a distance matrix and from that get a guide tree to make a phylogeny. Atleast thats how the method goes.
BTW i found this. Hope it helps a little. http://scikit-bio.org/docs/0.2.3/generated/skbio.io.phylip.html
Thanks to the both of you for your replies. I don't really have to use Biopython but I got intrigued by this library because I'm a huge Python fan. I'll try to explain what we have to do for our project.
This group project is about building three phylogenies of Sars-CoV-2 using different techniques and is divided as follows:
a) Using BLAST, perform a pairwise comparison of sequences and use such data to build a Phylogeny using Phylip;
b) Using ClustalW2, perform multiple sequence alignment and build a phylogeny with Phylip and compare it with the one in (a);
c) Using EPSim, try to compute pairwise distances and build a phylogeny with Phylip and compare it with the one in (a) and (b);
d) Using distances of point (c), cluster sequences together and compare results with (a) and (b). A cluster should be contained in a subtree.
I'm in charge of point (a). I immediately realized that pairwise alignment is possible only on the web UI of BLAST at https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&PROG_DEF=blastn&BLAST_PROG_DEF=blastn&BLAST_SPEC=GlobalAln&LINK_LOC=BlastHomeLink and it is based on the Needleman-Wunsch algorithm. Since it's not possible to use the web UI to make several pairwise alignments, I decided to move to other tools. That's how I stumbled on biopython.
Seems like Biopython isn't gonna work for what I have to do. @gayachit I found something similar to the distance matrix and guide tree here https://link.springer.com/content/pdf/10.1007/978-3-642-01970-8_96.pdf. In page 3, they are created as steps 1 and 2 of the ClustalW algorithm...which is the part assigned to my collaborator.
So to sum up: is it really possible to build a phylogeny only by repeated pairwise comparisons? If so, what's the best software for that particular task?
Thank you very much!
Sounds like you need a distance method like neighbor joining method. You could use MEGA tool: https://www.megasoftware.net/ It states in the manual that In distance methods, a pairwise evolutionary distance is computed for all species or OTUs to be studied, and a phylogenetic tree is constructed by certain principles and algorithms.
Gayachit thank you very much for your help. I have a couple of questions:
a) Can I ask you to point out in the manual the part where it is specified that pairwise distance is used? I will probably need it for future reference
b) Among the reputation of different bioinformatics software, is MEGA considered to be reliable? Is it used in the academia? Do you also have some other suggestions for software that can perform Neighbor Joining Phylogeny?
For 1: https://www.megasoftware.net/mega1_manual/Phylogeny.html#5-1 under tree building methods
For 2: MEGA is used by many for phylogenetic analysis and has been cited in multiple papers. There are a number of tools creating phylogenetic trees. You need to google about the methods they use.
Thank you very much for your help. You really helped a lot :)
Have a good day!