Entering edit mode

3.6 years ago
Morris_Chair
▴
350
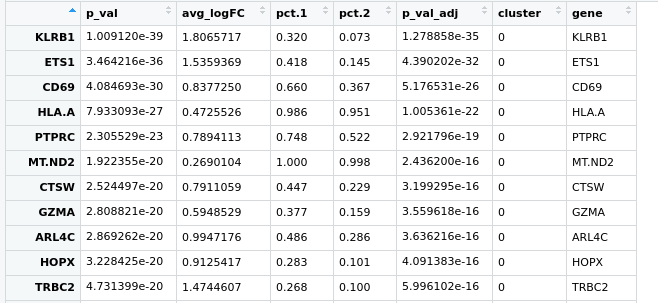
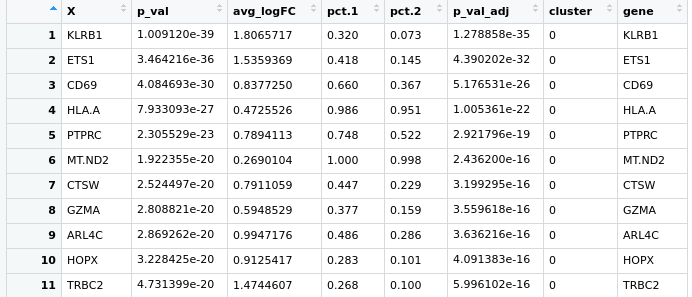
Hello everyone, I have a little problem with Seurat pipeline, I created a heatmap based on the top expressed genes in each cluster using the following code:
pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
here is the pic https://ibb.co/zfWD1Wh
it's not clear to me how can I select more genes to represent in this heatmap, if I do more the n=100 doesn't change anything. I have a gene of interest in the cluster 5 which based on the avi_LogFC, is at the position 160th and I'd like to show it here .. do you guys have any suggestion?
thanks for help :)
Hmm could you share the full code you're using to generate the plot? Does
pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)feed directly into the plot, or is this supposed to updatepbmc.markersfirst? (Themagrittrpipe is uni-directional here, so it wouldn't updatepbmc.markerswith filtering criteria here.) As an aside, I would recommend replacingtop_n()withslice_max()orslice_min()as appropriate.