
Hi there,
I want to use the SCANB-Datasets (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81540) for classifying samples into PAM50 breast cancer subtypes. However, the dataset is only available as FPKM-log2-transformed data (only the very original expression data is also available, but I would like to avoid all these additional preprocessing steps they already carried out).
FPKM is not suitable for cross-sample comparisons. What normalization can I put on top of the data to achieve cross-sample comparability? I guess I need to re-log2-transform the data first to then apply another normalization strategy?
Is this a valid strategy at all? Should I try to back-transform the FPKM values to the original counts to start a fresh normalization?
I appreciate your help with this!
Best, Cindy

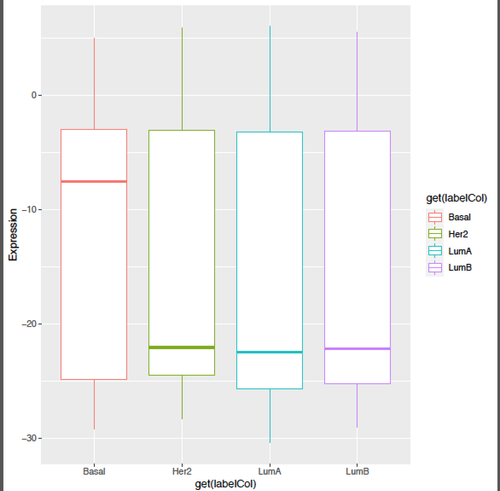
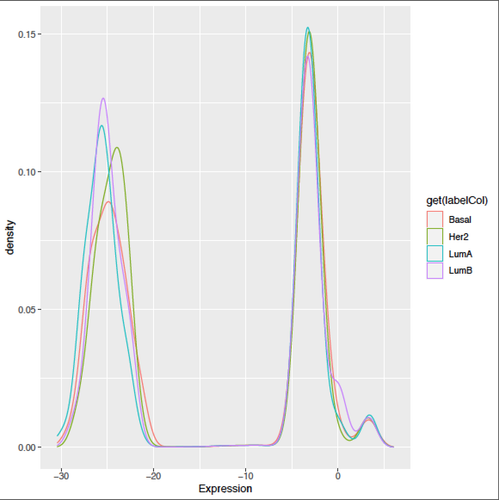
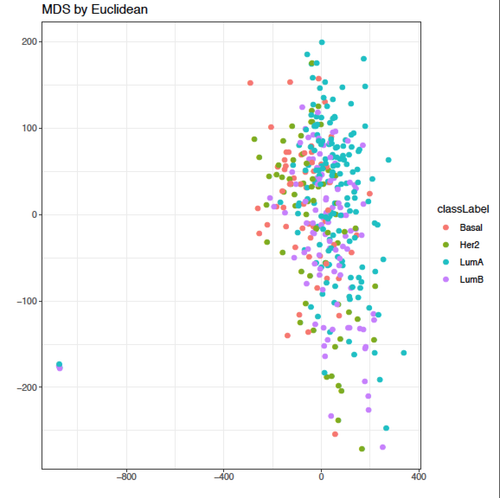
Kevin, thanks a lot for your helpful input!
I had thought of the same regarding FPKM-UQ, but when researching about the TCGA method, it looked as if the upper quartile normalisation was carried out during the FPKM normalisation (see here: https://docs.gdc.cancer.gov/Encyclopedia/pages/HTSeq-FPKM-UQ/):
Besides that, I had not found any other resource stating that it is ok to carry out upper quartile normalization on top of FPKM (so I was a bit unsure about that).
Best
Cindy
Why not just convert FPKMs to TPMs?
No, raw counts are not available (that's exactly my problem). There is a file called "GSE81538_gene_expression_405_transformed.csv" but it does not look like raw counts to me, so I have no idea at what point in the analysis this file was generated.
According to the study, that is how they preprocessed the data:
Here is an excerpt from GSE81538_gene_expression_405_transformed.csv:
Best,
Cindy