Entering edit mode

5.7 years ago
anoops
▴
40
Hello,
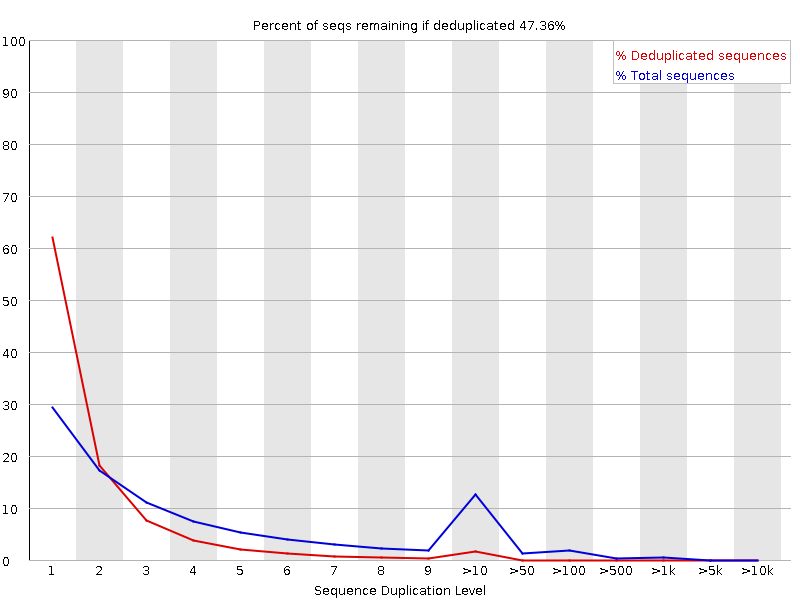
I am trying to convert a batch of BAM files to FASTQs. I started out testing SAMTOOLS (collate/bam2fq) and PICARD (SAMTOFATQ). On the outset the numbers seemed OK but the statistics suggests that the SAMTOOLS out has twice the amount of duplicates as the Picard out.
Has anyone experienced this? I am not sure if it is a samtools problem or I am not comprehending the QC stats.
Any advice/recommendation/comments are welcome.
Thanks!
PS: In both cases I am outputting both first end of the pair and the second end of the pair as separate files.
UPDATED: The commands used were:
Samtools
samtools collate -o name-collate.bam sample.bam
samtools fastq -1 sample_1.fastq.gz -2 sample_2.fastq.gz -0 sample_0.fastq.gz name-collate.bam
Picard
java -Xmx2g -jar picard.jar SamToFastq I=sample.bam FASTQ=sample_1p.fastq.gz SECOND_END_FASTQ=sample_2p.fastq.gz UNPAIRED_FASTQ=sample_0p.fastq.gz
Fasqc check
fastqc -o fastqc_out/ sample_1p.fastq.gz
Picard QC
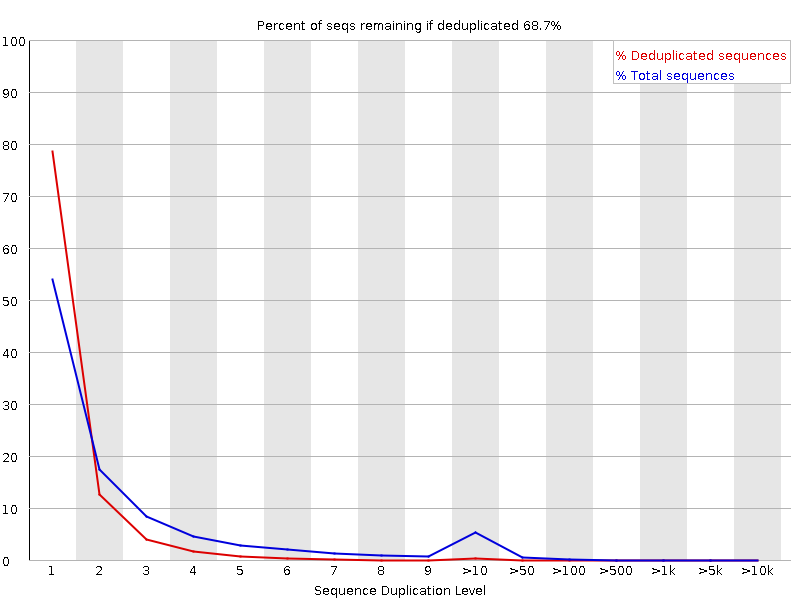
Samtools QC

It would be a big help if you could provide the command lines used
I didn't include them because they were default. They are included now. Thanks in advance!!!
FYI, you do not need collate. A simple sort by name with the
-noption ofsamtools sortwill "restore" the read order as it was obtained from the sequencer, so pretty much random. This you can directly pipe into samtools fastq:Good tip, thanks ATpoint
Actually, collate is faster than
samtools sort, and works fine for your purpose. Fromman samtools:Hello anoops!
It appears that your post has been cross-posted to another site: http://seqanswers.com/forums/showthread.php?t=83934
This is typically not recommended as it runs the risk of annoying people in both communities.
Sorry, did not realize. Will keep in mind.