Entering edit mode

2.7 years ago
Pratik
★
1.0k
Hello Biostars Community,
How would I plot biological replicates on a genomic features plot for methylated array?
Should I plot the mean from all the samples for each probe? or would it be better to plot each biological replicate separately? Hoping there is a way to combine them so it's concise in a way?
I want to plot figures like this: What cut-offs to use for Genomic Features Pie Chart for Methylation Array Data?

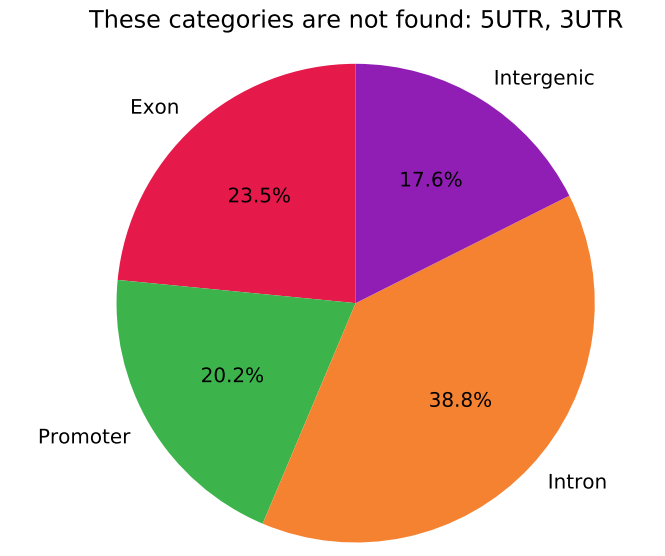
Thank you in advance : )

I really appreciate you going above and beyond to help me Papyrus! I have a couple different groups and each group has more than a couple biological replicates. Is mean/average a good way to pool together the biological replicates? Or is there a better way?
Thank you again. Really looking forward to your response!
You can take the mean/average to pool the replicates, and it is OK. Nonetheless, you have another option which does not involve losing/pooling information across the replicates. As I said, if you directly input all the replicate points (without taking the mean) into the boxplot/violin plots, the results should be pretty similar, because you have many CpGs and most are correlated between your replicates. You can check the two approaches.
Try this example in R: