
Hello,
I'm getting an error from DESeq2 that insists I have duplicates somewhere in my data, but I don't think I do.
I loaded transcript data from kallisto using tximport(). I wanted HUGO gene-wise counts, not transcript counts, so I aggregated these and fed the result into a DESeq object using DESeqDataSetFromMatrix. I double-checked to make sure that neither the rownames or column names in this count matrix were duplicated, but DESeq is still throwing an error.
Any ideas as to what might be the source of the issue, or what exactly DESeq is pointing to when it says "names(x)"?
Thanks for your help!
Code:
# pull count data out of tximport-created data frame for manipulation
hugoCrunch <- data.frame(assay(dds_kal))
# use my own function to add HUGO names using bioMart
hugoCrunch <- addHUGOnames(hugoCrunch, 'hsapiens_gene_ensembl', 'ensembl_transcript_id')
## add back into original data frame
hugoCrunch_data <- hugoCrunch[,-c(52:54)]
hugoCrunch_names <- hugoCrunch$Gene # these are the HUGO genes produced with addHUGOnames. There are duplicates here
# add genes together
hugoCrunch_crunched <- aggregate(hugoCrunch_data[], by = list(hugoCrunch_names), FUN = sum)
row.names(hugoCrunch_crunched) <- hugoCrunch_crunched[,1]
# row names are now HUGO genes, which are not duplicated
# remove row names and the NA gene counts
hugoCrunch_crunched <- hugoCrunch_crunched[-1,-1]
# ensure everything is unique
rownames(hugoCrunch_crunched) = make.names(rownames(hugoCrunch_crunched), unique=TRUE)
colnames(hugoCrunch_crunched) = make.names(colnames(hugoCrunch_crunched), unique=TRUE)
# put data back into a DESeq object
dds_kal_agg <- DESeqDataSetFromMatrix(countData = hugoCrunch_crunched,
colData = sampleTable_plus,
design= ~ Genotype)
# do sampleTable_plus IDs have duplicates in the first row (DESeq ID row)?
any(duplicated(sampleTable_plus[,1])
FALSE
# DESeq
dds_deseq <- DESeq(dds_kal_agg)
Error in
rownames<-(*tmp*, value = names(x)) : duplicate rownames not allowed
SessionInfo():
R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] gdtools_0.1.7 ggrepel_0.8.0 ggplot2_3.0.0 limma_3.36.5 pheatmap_1.0.10
[6] biomaRt_2.36.1 DESeqAid_0.2 DESeq2_1.20.0 SummarizedExperiment_1.10.1 DelayedArray_0.6.6
[11] BiocParallel_1.14.2 matrixStats_0.54.0 Biobase_2.40.0 GenomicRanges_1.32.7 GenomeInfoDb_1.16.0
[16] IRanges_2.14.12 S4Vectors_0.18.3 BiocGenerics_0.26.0
loaded via a namespace (and not attached):
[1] httr_1.3.1 bit64_0.9-7 splines_3.5.1 Formula_1.2-3 assertthat_0.2.0 latticeExtra_0.6-28
[7] blob_1.1.1 GenomeInfoDbData_1.1.0 progress_1.2.0 yaml_2.2.0 pillar_1.3.0 RSQLite_2.1.1
[13] backports_1.1.2 lattice_0.20-35 glue_1.3.0 digest_0.6.17 RColorBrewer_1.1-2 XVector_0.20.0
[19] checkmate_1.8.5 colorspace_1.3-2 htmltools_0.3.6 Matrix_1.2-14 plyr_1.8.4 XML_3.98-1.16
[25] pkgconfig_2.0.2 genefilter_1.62.0 zlibbioc_1.26.0 purrr_0.2.5 xtable_1.8-3 scales_1.0.0
[31] svglite_1.2.1 htmlTable_1.12 tibble_1.4.2 annotate_1.58.0 withr_2.1.2 nnet_7.3-12
[37] lazyeval_0.2.1 survival_2.42-6 magrittr_1.5 crayon_1.3.4 memoise_1.1.0 foreign_0.8-71
[43] prettyunits_1.0.2 tools_3.5.1 data.table_1.11.4 hms_0.4.2 stringr_1.3.1 locfit_1.5-9.1
[49] munsell_0.5.0 cluster_2.0.7-1 AnnotationDbi_1.42.1 bindrcpp_0.2.2 compiler_3.5.1 rlang_0.2.2
[55] grid_3.5.1 RCurl_1.95-4.11 rstudioapi_0.7 htmlwidgets_1.3 labeling_0.3 bitops_1.0-6
[61] base64enc_0.1-3 gtable_0.2.0 curl_3.2 DBI_1.0.0 R6_2.2.2 gridExtra_2.3
[67] knitr_1.20 dplyr_0.7.6 bit_1.1-14 bindr_0.1.1 Hmisc_4.1-1 stringi_1.2.4
[73] Rcpp_0.12.19 geneplotter_1.58.0 rpart_4.1-13 acepack_1.4.1 tidyselect_0.2.4
EDIT
Ah, of course I found a relevant post as soon as I post my own: https://support.bioconductor.org/p/113718/
My issue was solved once I changed the DESeq line to:
# DESeq
dds_deseq <- DESeq(dds_kal_agg, minRep=Inf)
I'm not entirely sure how this worked. When I follow the code on this website, it does look like my data have a few outliers: https://www.bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
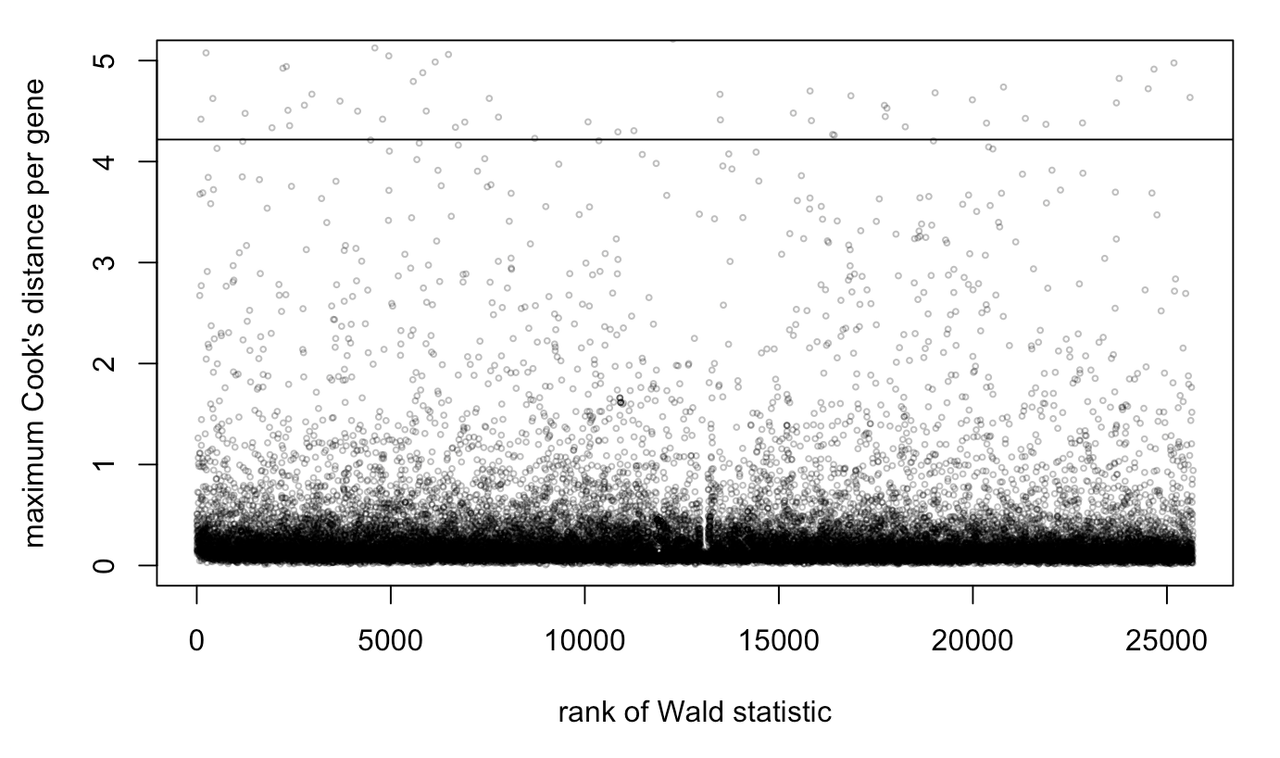

Thank you for taking a look at this!
Thanks for checking in and following up, Michael!