
Hi,
In single-cell RNA-seq it is widely used filtering by some metrics as a way of rough QC to remove dead cells:
- number of Counts
- number of Features
- % mitochondrial reads
- % ribosomal reads
- % unspliced reads
All these are used as proxy of empty droplets, doublets (excess of Counts and Features), dead cells, cells with broken membrane leaking out their cytoplasm, etc.
However, most if not all of these features have been shown to also reflect actual biological differences between cell types. For example, in this paper they discuss (and later identify) how metabolically active cell types have with high % mitochondrial reads. There are cell-type-dependent differences in the number of counts and features.
I've also found online some talk about cells with a secretory phenotype having higher % ribosomal genes. Although, [citation needed] for most of those claims, in figure 2 E&F of the ddqc method reanalysis shows B and T-cells having very high % ribosomal:

As for the % of unspliced reads, one can imagine many biological processes causing differences (e.g., multi-nucleation, changes in cell size). However, the standard practice is either to ignore it (high Malat1 expression in cells stressed by tissue dissociation), or to use intronic/Malat1 expression as a proxy of loss of nucleus/empty droplets made of ambient cytosol remains
High Malat-1 expression in single cell data
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-11015-5
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-11015-5/tables/1
Or the effect of low QC cells with no-nucleic reads:
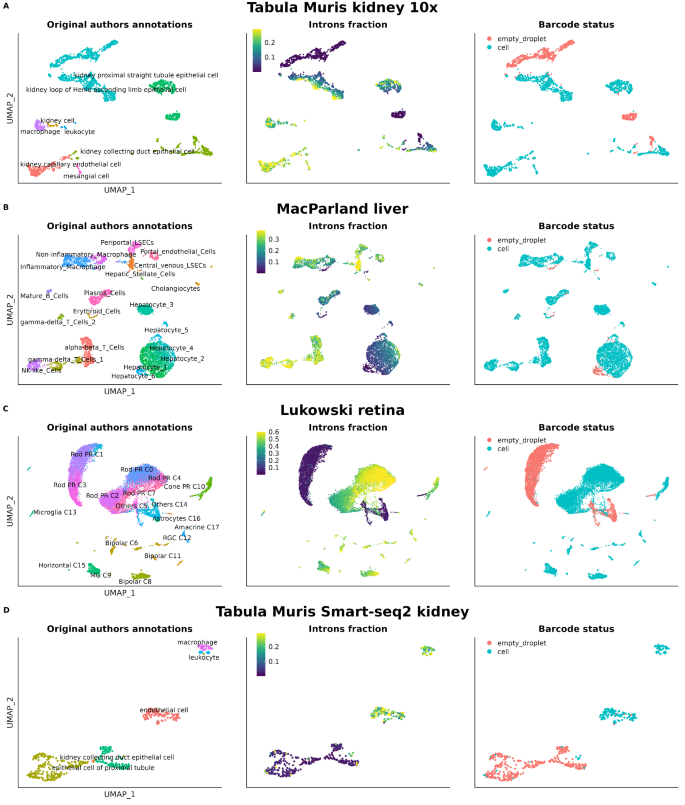
IMHO, even if these metrics end up being markers of low-quality cells, there is still value in identifying common characteristics of these cells. For example, in disease models and/or stressed tissues, where certain cell subtypes are enriched in low-quality cells but still show clear subtype-specific markers that make sense in the context of the tissue/disease.
My question is... is there any resource where this has been comprehensively studied? I'm looking for something that can be used as reference. It is a bit scary to include data in my analyses that a reviewer might deem too low quality and forces me to re-run the whole thing from step one during manuscript revision.
Also, do anyone know of good references about the relationship between secretory phenotypes and high % ribosomal reads? Or about the association between very low or very high intronic fraction with biological features?
Thanks in advance

Thanks!
I'm basically doing the same as you. I have already annotated my clusters and I was re-checking the QC metrics and saw some cells with uncomfortably-low QC metrics, so I wanted to revamp the whole thing.
My biggest concern is that, the current data I am analyzing is a disease model whose tissue requires very strong dissociation (lots of low QC cells). However, it is also full of "diseased" cell types that we are interested in. Those diseased cell clusters have concerning QC values, but are expressing specific markers known in the literature. Thus, they aren't just poor quality/dying cells misclustered from a different cell type.
After having done a first pass of QC (+ ambient and doublet correction/removal), this is an example of the QC I have:
Cluster #3 (~700 cells) looks highly suspicious. High %mt and % unspliced + low nCounts suggests (as per this table ) damaged or dying cells. However, when looking at these cells in detail (see figure below), there isn't a clear correlation between %mt and %unspliced. This makes me doubt. Are these (more) damaged cells, or are these a population with both naturally high %mt and %unspliced reads? I'll have to check back to our tissue experts to see what do they know (and have previously validated) about these cells. They might have been deemed as senescent-like in the past? This study suggests that ribosomal protein synthesis is impaired in senescent cells, which would match these extremely low %ribo levels. And this other study suggests an increase in mitochondrial mass in senescent cells, which could lead to high %mt.
Other high % unspliced clusters like #14 and #17 have very low %mt and nCounts, so they don't match the description for "bare nucleus" cells.
When looking at some of these clusters in detail, I cannot see (at least as a first glance) clear trends pointing to one or another direction. Maybe #14 could be split into 2 groups, with one of them lower quality.
I annotated cluster both #17 and #18 as macrophages. Some studies have detected populations of silent vs activated macrophages, having different number of nCounts, although there is no information about the % unspliced reads. The low %ribo of #17 could also align with a silent state according to this study on ribosomal activation in immune cells
Is looking into % unspliced reads really useful, beyond an initial check for extremely low levels? (regardless Malat1 expression, all my cells have >1.5% unspliced reads). Or is focusing on this (and %mt or %ribo) just overanalyzing noisy data that leads nowhere?
I think I'll err on the side of using relatively lax cluster-specific QC thresholds. I'll limit my QC to "clean up outliers" in each cluster as much as possible. And then try to find biological explanations/justifications for those clusters with consistently altered metrics.
Thanks again for your insights.
Yeah, those clusters are suspicious. But if you feel the biology explains them appropriately, then lean on that knowledge. Just be prepared to defend it. I'd still check Malat1 expression to see how it looks. As mentioned, I've found (un)spliced read% a difficult metric to do anything with beyond removing the very low % clusters, but I just use Malat1 for that since the range is more consistent.
If you think senescence may be driving these metrics, take a look at some senescence markers (low LMNB1, high GLB1) or hunt around the literature for senescence-associated genesets to score in each cell.