Entering edit mode

10.8 years ago
Nikleotide
▴
130
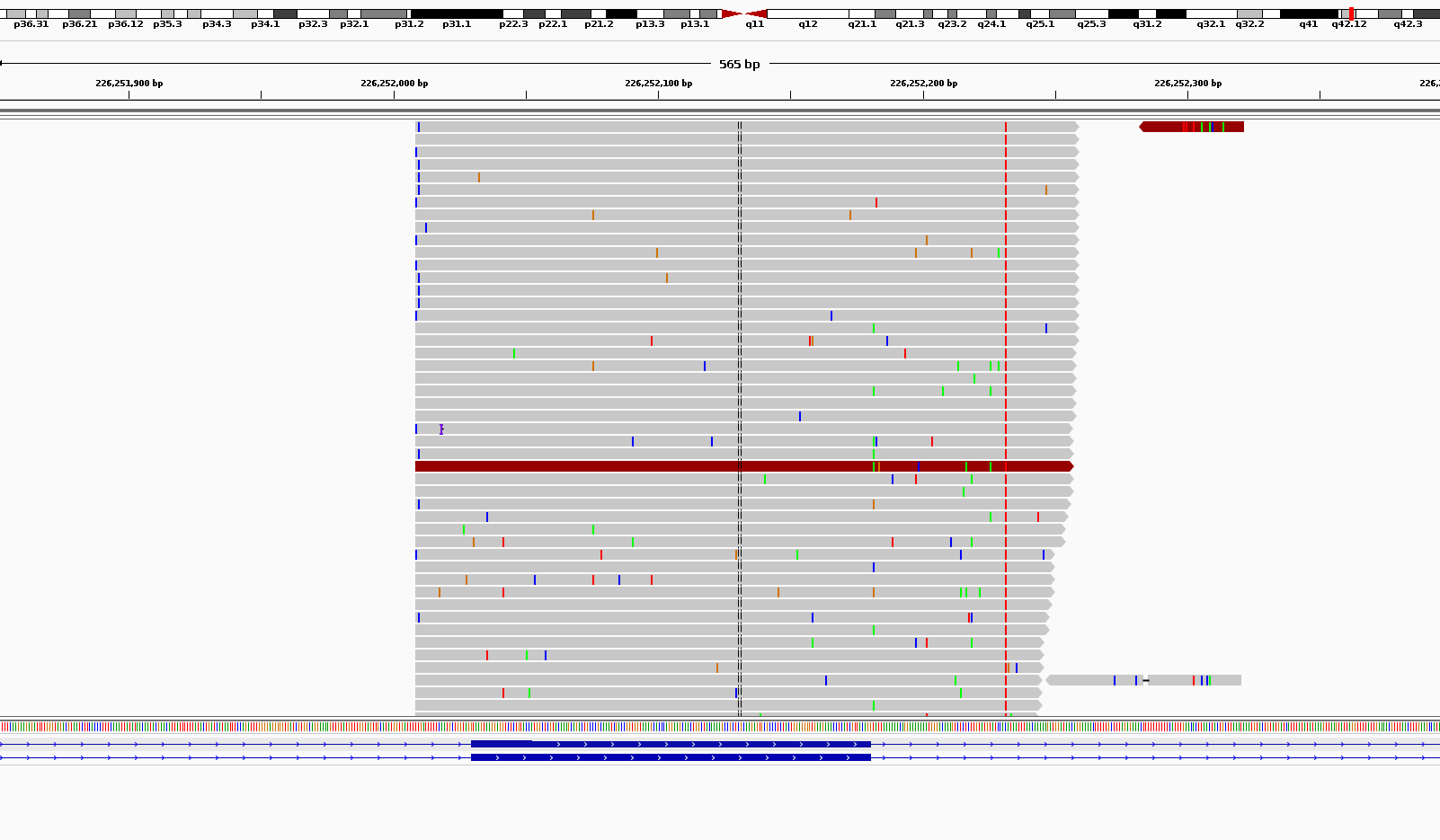
I am aligning MiSeq deep sequencing targeted amplicons to human genome (paired ended).
The resulting alignments look noisy. Since we are trying to fish for very low frequency variants, is there any way to count for the noises? Like how to calculate error rate in MiSeq bam files and how to consider them when calculating allele frequencies.
Any input will be appreciated.
Here is an example of the noise I am referring to.