Entering edit mode

8.3 years ago
cpak1981
▴
140
HISAT2 manuals says for:
For paired-end reads, use either FR or RF.
With this option being used, every read alignment will have an XS attribute tag:
'+' means a read belongs to a transcript on '+' strand of genome.
'-' means a read belongs to a transcript on '-' strand of genome.`
Why does it matter whether I specify FR or RF if every read is tagged with an attribute + or -?
---Edit based on current answer---
Follow up Qs:
Is + and - defined as the 5'-3' and 3'-5' DNA (genomic) strands, respectively? Is FR defined as read-1 is the reverse-complement of the annotated gene-orientation? Therefore, FR PE reads may come from the + or - strand?

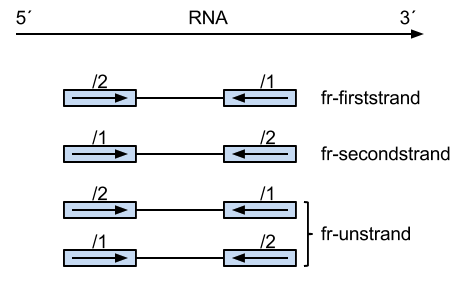

Thanks for the reply. Just so I understand: Is
+and-defined as the5'-3'and3'-5'DNA (genomic) strands, respectively? IsFR(with PE reads) defined asthe first-read is the reverse-complement of the annotated gene-orientation? Therefore,FRPE reads may come from+or-strand?