
Hi everybody,
I am new to the field of bioinformatics, sequence alignment and variant calling, but I have recently been hired to analyze whole genome sequences (paired-end) of grape vine accessions. To start, I have run FastQC on my fastq files and aggregated results using MultiQC. The quality scores were very good for all files (not shown) but I got worried with two other metrics.
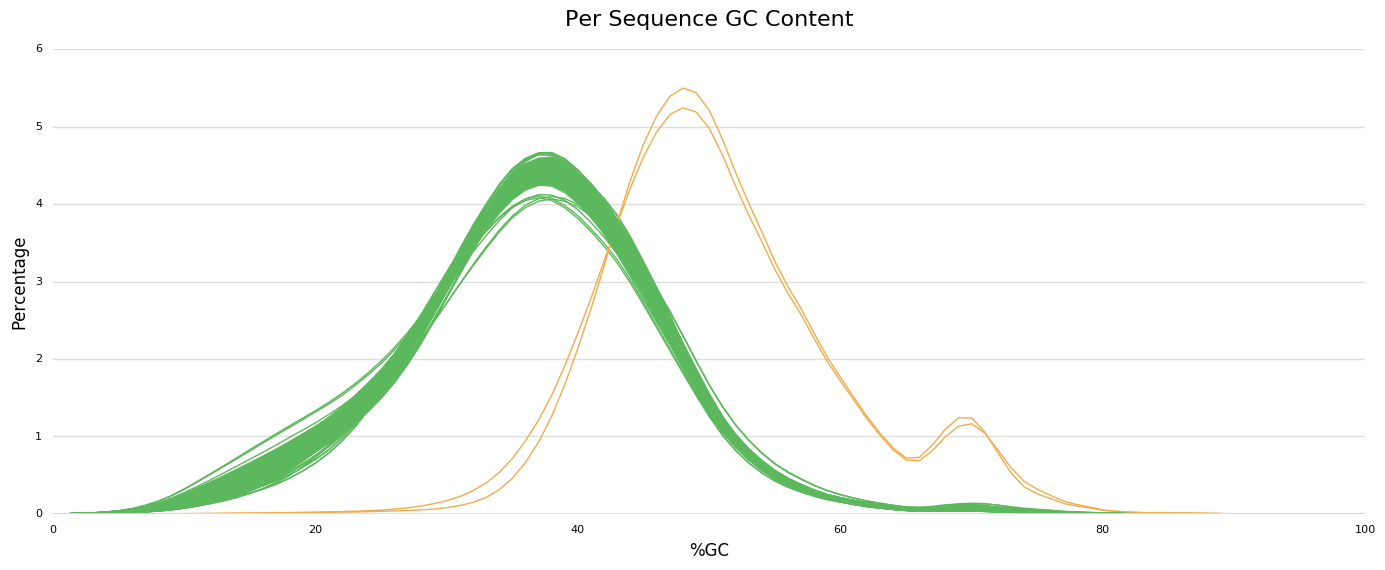
First, the GC content is expected around 35% and looks OK for most individuals, except for one individual for which the GC content is around 50% and has a small peak around 67-70%. Interestingly, this second peak could be observed for most individuals while being very flattened. This not clear to me if this is the result of a contamination of anything else?
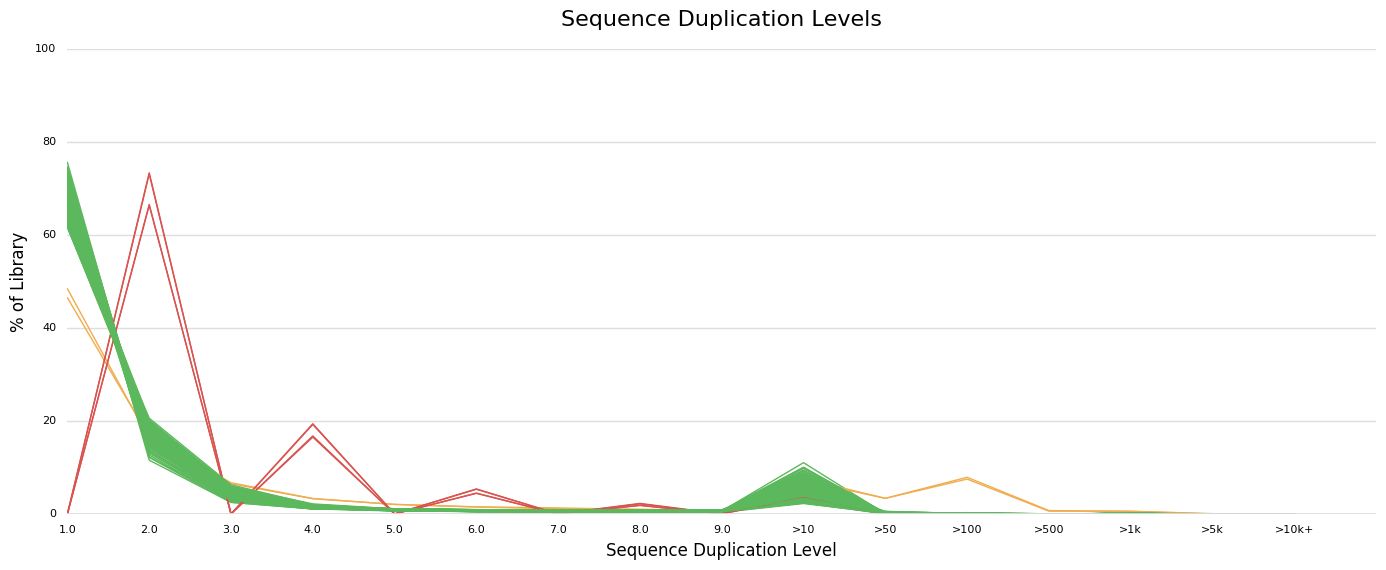
Secondly, the sequence duplication level looks OK for most individuals, except for three including the one identified based on GC content (in orange) and two other individuals (in red). The pattern looks very weird for these last two individuals as the duplication level is non-null only for even numbers (2, 4, 6...). I wonder how this is possible?
If anyone could help me interpret these results, that would be very nice of you. Also, I could use some guidelines on how to deal with those files (discard, contact sequencing company, run specific software...) Thanks for your help!
Those two samples look way too different than others. You should check to make sure they are not contaminants before you do anything else.
Is this plain genome sequencing data?
Please check this blog post by authors of FastQC, if you have not seen it :
https://sequencing.qcfail.com/articles/libraries-can-contain-technical-duplication/
Thanks for the quick reply and for the blog post!
This is indeed plain genome sequencing data.
I agree that the spotted samples are suspicious. My feeling is that the genotype identified based on GC content is contaminated. Could give me advice on how I could check this? or is it this observation the actual check leading to discard the files?
Regarding the two others genotypes identified based on sequence duplication level, I am wondering whether this "systematic" duplication of all reads can be dealt with by running standard variant calling pipeline (MarkDuplicates). The assumption is that the high duplication level is just a technical artifact whose main consequence is a lower depth for calling variant after removing duplicates Please tell me if that makes sense or if that's not reasonable.
Not sure what you mean by this? I am thinking that there may be no grape in those two samples at all based on their profiles.
Markdups will take care of PCR duplicates.