
Hi Everyone,
Background: I am working on a project where I want to predict putative short transcription regulatory motifs (~20 DNA bp). I do not typically work with sequences this short nor do I have a great sense on how people tend to predict (if at all) short DNA motifs. Nonetheless, my inclination is that HMMER (I am using v3.1b2) might be suitable for the task. To do this, I generated a multiple sequence alignment using a combination of sequences from previously published work and some sequences a colleague provided me. The alignment indicates a conserved CTG feature (mean frequencies 83%,100%,83%), followed by a moderately conserved CAG feature (mean frequencies 50%, 50%, and 75%, respectively), and last a more conserved CAG feature (mean frequencies 92%,75%,92%)--the CTG and second CAG feature were consistent with what was previously reported for this particular transcription regulatory motif.
Issue: My issue is that after generating the HMM profile using hmmbuild (default parameters), I try predicting the motif using nhmmer (default parameters) on the original DNA sequences used to generate the HMM profile--the idea being that all (or most) of the sequences should return as positive hits. As it turns out, none of the sequences return as positive hits. Inspecting the output file suggests that the forward parsing filter removes most sequences; however, utilizing the --max flag with nhmmer (which to my understanding removes the heuristic filters and returns everything meeting the e-value threshold) yields only few hits with unimpressive E-values (~0.1 to 1).
Question: My question is twofold. Can nhmmer feasibly predict DNA sequence motifs as short as 20 bp? If my understanding of HMMER is correct, the query sequence significance is determined based on how a HMM profile aligns with the query sequence with respect to a null model (I think the DNA frequencies are based on Swiss-prot genes). I can imagine since the conserved regions are so short, the query sequence and the random model are too comparable in likelihoods to confidently return a positive match. This I do not know. If nhmmer is theoretically sensitive enough to detect such a short DNA motif, how can I alter the HMMER parameters to improve my predictions.?
Thank you for any input.


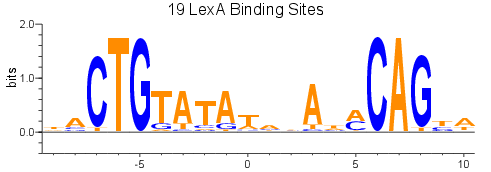
Thank you for the quick response and suggestion. The MEME suite looks promising and I will explore its implementation!