I was wondering why during sequencing process the fragment (insert size + adapters. See fig.) is not sequenced completely but, starting from the adapters, just the first part.
Can someone explain it since I can not retrieve information anywhere ?
In the image you have above insert size is a certain number of base pairs. For most common types of libraries that would be in 300-500 bp range.
Since the longest read lengths (without doing a non-standard set up, like running a 600 bp paired end kit as a single end run) Illumina supports at this time are 300 bp you generally will not see any adapter read-through (where a read will cross the entire insert and then go into read 2 adapter). In cases where the insert size is smaller than the number of cycles sequenced this will be evident (insert sizes are never exactly identical, there is a normal distribution of sizes with tails).
Illumina sequencing primer sits at the edge of adapter so the first base read 1 starts with is your insert.
Thanks for answering, but I can not still understand why it is left a "not sequenced" segment, I mean, when the library is prepared why are chosen fragments long enough that the read 1 and read 2 do not overlap? I thought read 1 and 2 would sequence the same bases.
It is a matter of getting the best return for your money. Having fragments of that size allow you to sample wider swaths of genome more effectively. It also allows bridge amplification to work well, which is critical for Illumina sequencing. In many applications you are aligning to a reference. Using reads that do not overlap in middle allows them to be placed/mapped at a distance. One can thus infer the sequence of the entire fragment sampled by R1/R2 reads.
There are definitely applications where one wants Read1 and Read2 to overlap. Initially people used to do that since quality of illumina reads dropped later in sequencing cycles (in very early days) so having reads that overlapped gave an estimate of sequencing error. Because of the improved sequencing technology this kind of error checking is no longer needed or is of concern.
There is nothing that prevents one from making libraries (with shorter inserts) so Read 1/Read2 sequence the same bases and perfectly overlap. Many would consider that a waste of money.

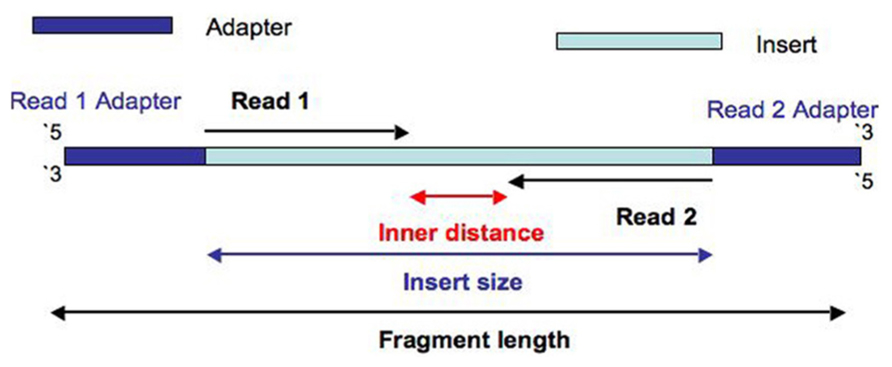

Thanks for answering, but I can not still understand why it is left a "not sequenced" segment, I mean, when the library is prepared why are chosen fragments long enough that the read 1 and read 2 do not overlap? I thought read 1 and 2 would sequence the same bases.
It is a matter of getting the best return for your money. Having fragments of that size allow you to sample wider swaths of genome more effectively. It also allows bridge amplification to work well, which is critical for Illumina sequencing. In many applications you are aligning to a reference. Using reads that do not overlap in middle allows them to be placed/mapped at a distance. One can thus infer the sequence of the entire fragment sampled by R1/R2 reads.
There are definitely applications where one wants Read1 and Read2 to overlap. Initially people used to do that since quality of illumina reads dropped later in sequencing cycles (in very early days) so having reads that overlapped gave an estimate of sequencing error. Because of the improved sequencing technology this kind of error checking is no longer needed or is of concern.
There is nothing that prevents one from making libraries (with shorter inserts) so Read 1/Read2 sequence the same bases and perfectly overlap. Many would consider that a waste of money.
Great, Thanks for helping!