
Hi everyone, I was wondering if someone can point me to a good explanation where i can better understand how the reads in my fastq files are created.
in the fastqc results from the data set I got, we assume that a lot of the adapter was sequenced (2. image). I'm not talking about read-thourgh, but the adapter somehow was sequenced. At least we think this is what we have.
I have tried to understand in which direction the reads in my fastq files are read. 
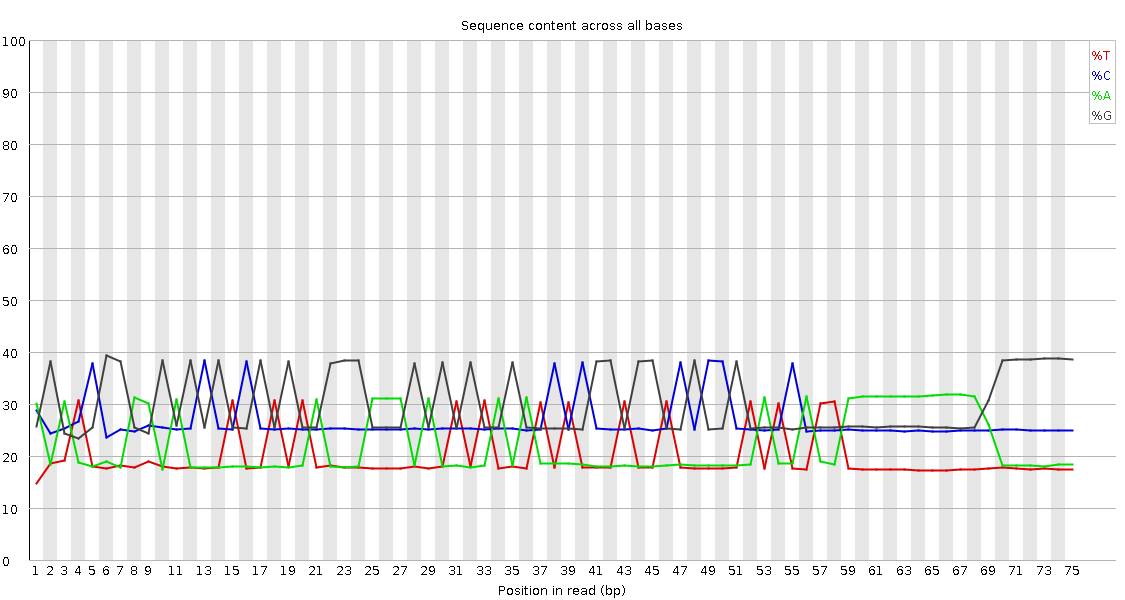

No, this should be a normal ChIP-Seq data from drosophila genome. There shouldn't be this kind of behavior. What i do know the the sequencing facility did was a paired-end 75nt (so 150nt in total). In previous projects which worked better, we got a read length of 42nt. This is why i think I need to trim it somehow, just not sure which side. So 3' would be the end of the read in the fastq file directionality. Am I correct?
Sequence data is always represented in 5' ---> 3' orientation. If you obtained data that is longer than what you expected you may not have to trim it down. It should still align fine as long as your insert size was > 75 bp. But if you must then you should trim the sequence at 3'-end of reads in both R1/R2 files.
I did this, only to get like 50% mapping in the Input and 12%(!) in the IPed samples. Even after filtering for possible adapters (Truseq) I still very low results. I still can't figure out what is going on here.
At times you may have bad libraries or bad sequencing data. You have no option but to start with reads that are not aligning and dig in. Take a sample do blast at NCBI. Start making sure that there was no contamination. These are indeed your data and go from there.