Entering edit mode

2.3 years ago
シン
▴
10
I am trying to do exactly the same pipeline for RNA-seq data process as the TCGA does. Usually when we ask a sequencing service, we can get a fastQ file. It contains sequence and read quality information. The alignment step comes next. However in the case of TCGA, as their pipeline suggested (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/). It seems that they used BAM files as one of the inputs. I was wondering why they used BAM files as inputs and how can I repeat what they did? In addition, why isn't there seem to be a adaptor trimming process?
1: 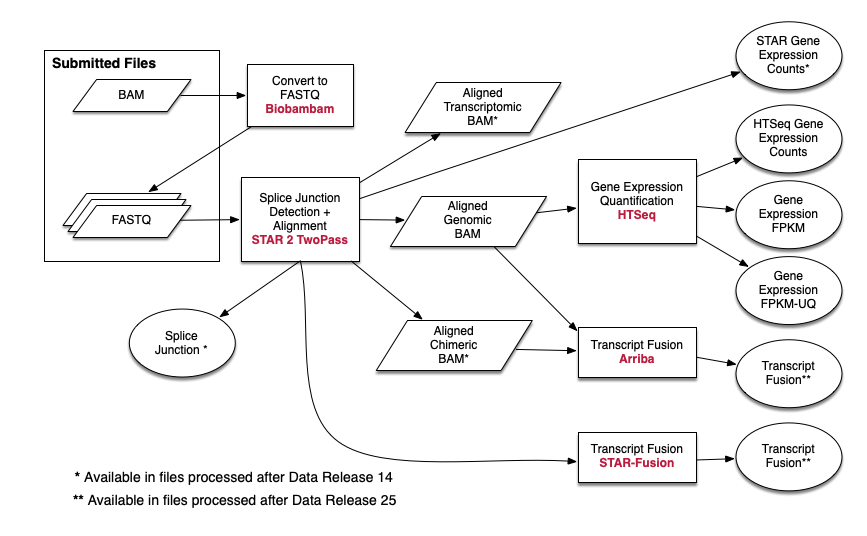 https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/


Thank you Delaney, these links answered many questions of mine.