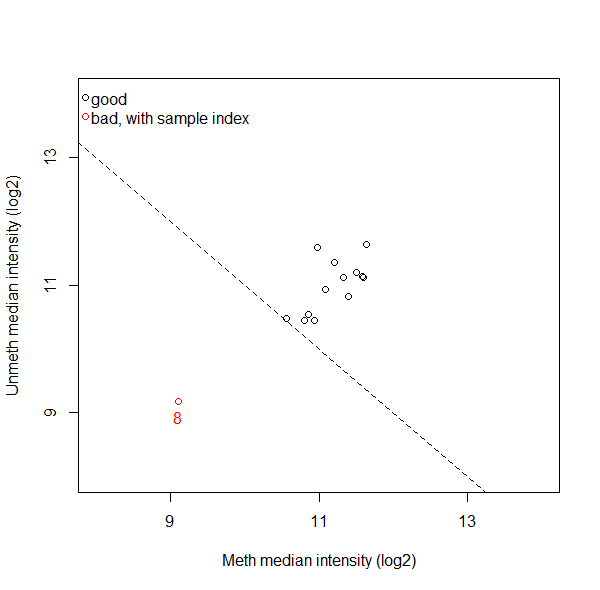I have a question concerning the sample quality of Illumina Infinium HumanMethylation arrays. I am analyzing a dataset consisting of 28 paired samples obtained from FFPE material of 14 patients. The samples are subdivided into two groups, whereby each group pair corresponds to one patient. The samples were distributed on the EPIC slides in a balanced fashion (i.e. block design). I'm analyzing the data using mostly using ChAMP, minfi and I will use Limma for differential methylation analysis of the paired samples. Sample quality was also assessed using shinyMethyl.
Unfortunately, some samples have an overall poorer quality, and I'm thinking about removing them from the analysis. However, since the cohort is small and the samples are paired, removing a sample means removing the whole sample pair, I'm a little reluctant to remove too many samples. Or is it possible to perform an unbalanced paired analysis with Limma?
As mentioned above I use different packages, all of which offer a wide range of quality metrics and plots. But neither in the user manuals nor in the literature have I found which is the most important criteria to exclude bad quality samples.
For instance, ChAMP calculates the fraction of failed positions per sample, which corresponds to the mean of the probes with a detection p-value below 0.01 and advises to manually exclude samples. I have one sample with a fraction of failed positions per sample of 0.0773193972, which substantially reduces the number of cg-probes in the analysis. Furthermore, the beta distribution deviates strongly from the other samples and the log2 intensities of the BISULFITE CONVERSION I and II QC probes are very low. So this is a quite obvious case, but what about more nuanced bad quality samples. What thresholds do you use for fraction of failed positions per sample and log2 intensity of BISULFITE CONVERSION QC probes?
Furthermore, minfi also has a function that plots the median of the unmethylated vs the methylated signal (see attached image). Are all samples labeled as bad not safe to use and should be deleted, or do you check whether a sample fails to pass more than one QC, or what do you usually do? And are there maybe better ways to assess which samples to exclude?