Entering edit mode

10.1 years ago
samsara
▴
630
I have RRBS fastq files. I used Bismark to perform methylation call. After methylation call I got M-bias plot shown below. The methylation rate of first three bases of 5 prime end is quite high. The actual methylation count and rate of first four position is shown below.
My questions are:
- Is the observed high methylation rate is because of end repair biases?
- In the literature It has been mentioned that it is common to have high methylation rate in 5' end, but how much is too much?
- First three bases of RRBS reads are either CGG or TGG depending on their methylation state. Is it good idea to chop off first 3 bases ? If yes, doesn't the removal of C (that retains original genomic methylation state) influence downstream analysis?
CpG context =========== position count methylated count unmethylated % methylation coverage 1 5000734 2489532 66.76 7490266 2 430 206 67.61 636 3 190 131 59.19 321 4 34174 79253 30.13 113427
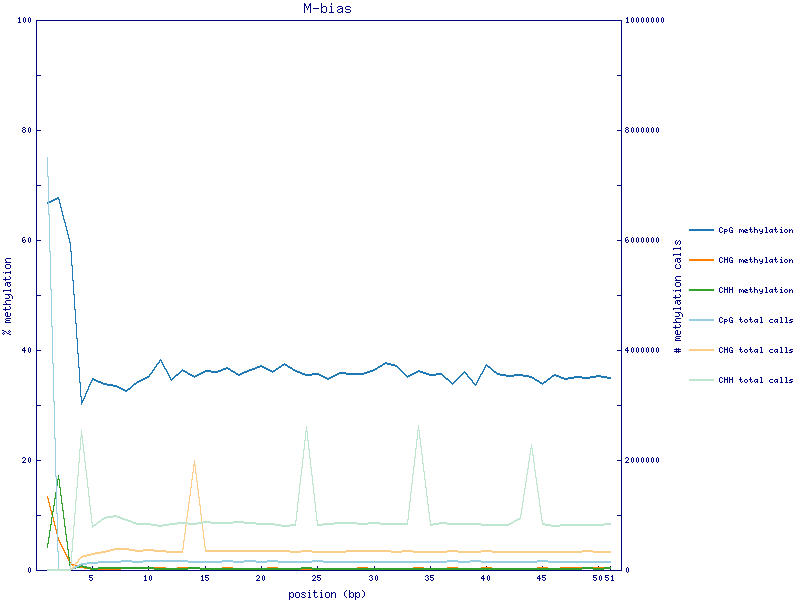

For RRBS, the majority of the reads start with CGG or TGG (at the 5'), and that's the MspI cutting sites left-over. For the M-bias plot, it plots methylation% in each base, there is a higher probability that the first base is methylated, other bases may even do not have a C, thus low methylation%. Does it make sense to trim the first three bases in this case?
Trim_galore with --rrbs option trimmed another 2bp from the 3' end to remove the filled (end-repair introduced) Cs (unmethylated)
I read from here http://www.bioinformatics.babraham.ac.uk/projects/bismark/RRBS_Guide.pdf
Thank you, Ming
I realize this is a very delayed followup, but I was hoping you might clarify #1. Shouldn't the end-repair impact the 2 bases at the end of the read, not the first 3 bases?
One would think so, yes, but for some reason the third base seems to be affected at least sometimes too. No clue why.
But still, shouldn't it be the 2 or 3 bases at the end, not the beginning of the read?
For example, end-repair is causing problems at the end of the molecule and thus, the beginning of R2 for WGBS is wrong. Or is that a separate issue?