
The previous tutorials in our GEO data mining series have shared the steps of downloading data, "GEO Data Mining (I) "Gene Expression Omnibus Data Mining (IA) – Quick and easy download of GEO data" and "GEO Data Mining (IB) - Downloading Sequence Read Archive raw data” and the steps of gene variance analysis, “GEO Data Mining (II) – Differential gene expression analysis and visualization”. In this article, we will share code necessary for performing pathway enrichment analysis of the extracted genes whose differential expression has been examined.
Using the earlier tutorials, we have already identified differentially expressed genes. However, that information is not fully complete. Because there are many gene categories, the biological significance of a set of differentially expressed genes may not always be obvious. Enrichment analysis examines whether a set of genes is more significant at a certain functional node compared to a random level and extending from simple annotation of a single gene to the analysis of grouped ness of multiple gene sets. Enrichment analysis can be useful for annotating the function of genes of interest, such as differentially expressed genes, and analyzing which gene set they are most enriched in, and what types of regulation they are involved in, thus affecting the occurrence of diseases. The most common enrichment methods used today are Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analyses to reveal the biological context represented by a class of genes.
GO term functional enrichment analysis
The Gene Ontology (GO) database is one of the most widely used gene pathway annotation systems. Its principle is simply to calculate whether differential genes involved in regulatory processes are significantly clustered in three levels: cellular component (CC), molecular function (MF), and biological process (BP).
“Cellular component” describes a specific cellular structure whereby the gene product performs its function. For example, a product protein may be localized in the nucleus or in the ribosome. Molecular function describes the activity of the gene product at the molecular level, such as catalysis or transport. Biological process describes a biological function with which a gene product is associated, or a large biological activity accomplished by the activity of multiple molecules. Examples include mitosis or purine metabolism. The code below will help you perform GO on your sample.
##import related R package
rm(list = ls())
load("step4_output.Rdata")
library(clusterProfiler)
library(dplyr)
library(ggplot2)
source("kegg_plot_function.R")
#(1)input data
gene_up = deg[deg$change == 'up','ENTREZID']
gene_down = deg[deg$change == 'down','ENTREZID']
gene_diff = c(gene_up,gene_down)
gene_all = deg[,'ENTREZID']
#(2) GO analysis, in three parts
library(org.Hs.eg.db)
if(T){
# Cellular component (CC)
ego_CC <- enrichGO(gene = gene_diff,
OrgDb= org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
# Biological process (BP).
ego_BP <- enrichGO(gene = gene_diff,
OrgDb= org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
# Molecular function (MF):
ego_MF <- enrichGO(gene = gene_diff,
OrgDb= org.Hs.eg.db,
ont = "MF",
pAdjustMethod = "BH",
minGSSize = 1,
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
save(ego_CC,ego_BP,ego_MF,file = "ego_GSE42872.Rdata")
}
load(file = "ego_GSE42872.Rdata")
#(3) Visualization
#Strip Chart
barplot(ego_CC,showCategory=20)
#bubble plot
dotplot(ego_CC)
geneList = deg$logFC
names(geneList)=deg$ENTREZID
geneList = sort(geneList,decreasing = T)
#(4) Demonstration of common genes in the top5 pathway
#Gene-Concept Network
cnetplot(ego_CC, categorySize="pvalue", foldChange=geneList,colorEdge = TRUE)
cnetplot(ego_CC, foldChange=geneList, circular = TRUE, colorEdge = TRUE)
#Enrichment Map
emapplot(ego_CC)
#(5) Demonstrate access relationships
goplot(ego_CC)
#(6)Heatmap-like functional classification
heatplot(ego_CC,foldChange = geneList)
pdf("heatplot.pdf",width = 14,height = 5)
heatplot(ego_CC,foldChange = geneList)
dev.off()
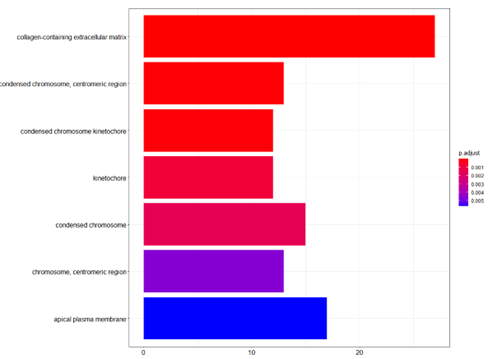
Histogram of GO Pathway enrichment analysis
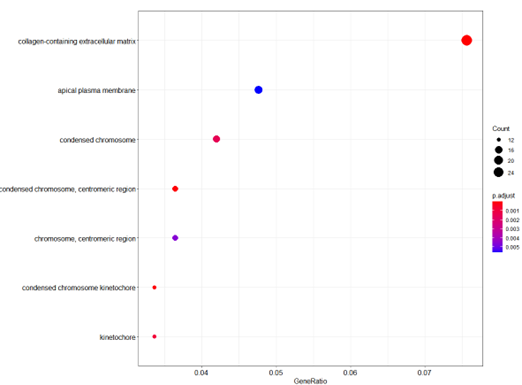
Bubble plot of GO Pathway enrichment analysis
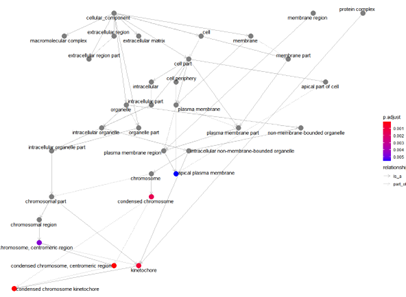
Access Relationships of GO Pathway enrichment analysis
KEGG enrichment analysis
KEGG pathway enrichment uses its own database for systematic analysis of gene function and genomic information, integrating information from genomics, biochemistry and systemic function, which helps us to study gene and expression information as a whole. Each KEGG pathway map contains a network of molecular interactions and reactions that link genes to pathways in gene products. Pathway analysis is useful for identifying the biological context of cellular and organismal metabolism as well as various other functions. The code below will help you perform GO on your sample.
#Up-regulation, down-regulation, difference, all genes
#(1) Input data
gene_up = deg[deg$change == 'up','ENTREZID']
gene_down = deg[deg$change == 'down','ENTREZID']
gene_diff = c(gene_up,gene_down)
gene_all = deg[,'ENTREZID']
#(2) Enrichment analysis of upregulated/downregulated/all differential genes
if(T){
kk.up <- enrichKEGG(gene = gene_up,
organism = 'hsa',
universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff = 0.9)
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa',
universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
kk.diff <- enrichKEGG(gene = gene_diff,
organism = 'hsa',
pvalueCutoff = 0.9)
save(kk.diff,kk.down,kk.up,file = "GSE21933kegg.Rdata")
}
load("GSE21933kegg.Rdata")
#(3) Extract the resulting data frame from the enrichment results
kegg_diff_dt <- kk.diff@result
#(4) Filter pathways by pvalue
down_kegg <- kk.down@result %>%
filter(pvalue<0.05) %>%
mutate(group=-1)
up_kegg <- kk.up@result %>%
filter(pvalue<0.05) %>%
mutate(group=1)
#(5) Visualization
g_kegg <- kegg_plot(up_kegg,down_kegg)
#g_kegg +scale_y_continuous(labels = c(20,15,10,5,0,5))
ggsave(g_kegg,filename = 'kegg_up_down.png')
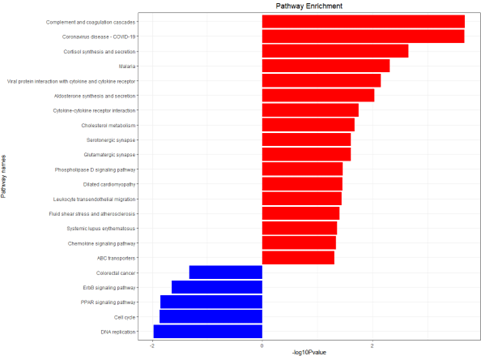
KEGG enrichment analysis histogram
Data mining of GEO database enables investigators to analyze differential expression, identify novel relationships or activities, extract significant conclusions, and build frameworks for new avenues of study. Investigators who are used to traditional work on coding genes can use the GEO datasets to enrich their studies with non-coding genes and explore DNA regulatory elements. If the sequencing depth from the GEO datasets is very deep, trans-shearing can be studied to mine potential circular RNAs. The original sequencing data can be analyzed from scratch to explore new genes. Therefore, data mining of GEO database enables investigators to explore gene functions and pathways expansively and in greater detail. The "GEO data mining" series has come to an end. In the future, we'll produce more insightful series. Remain tuned!
To get more information about Novogene, please visit our website: https://www.novogene.com/us-en/resources/blog/geo-data-mining-iii-gene-pathway-enrichment-analysis-with-complete-code-sharing/