
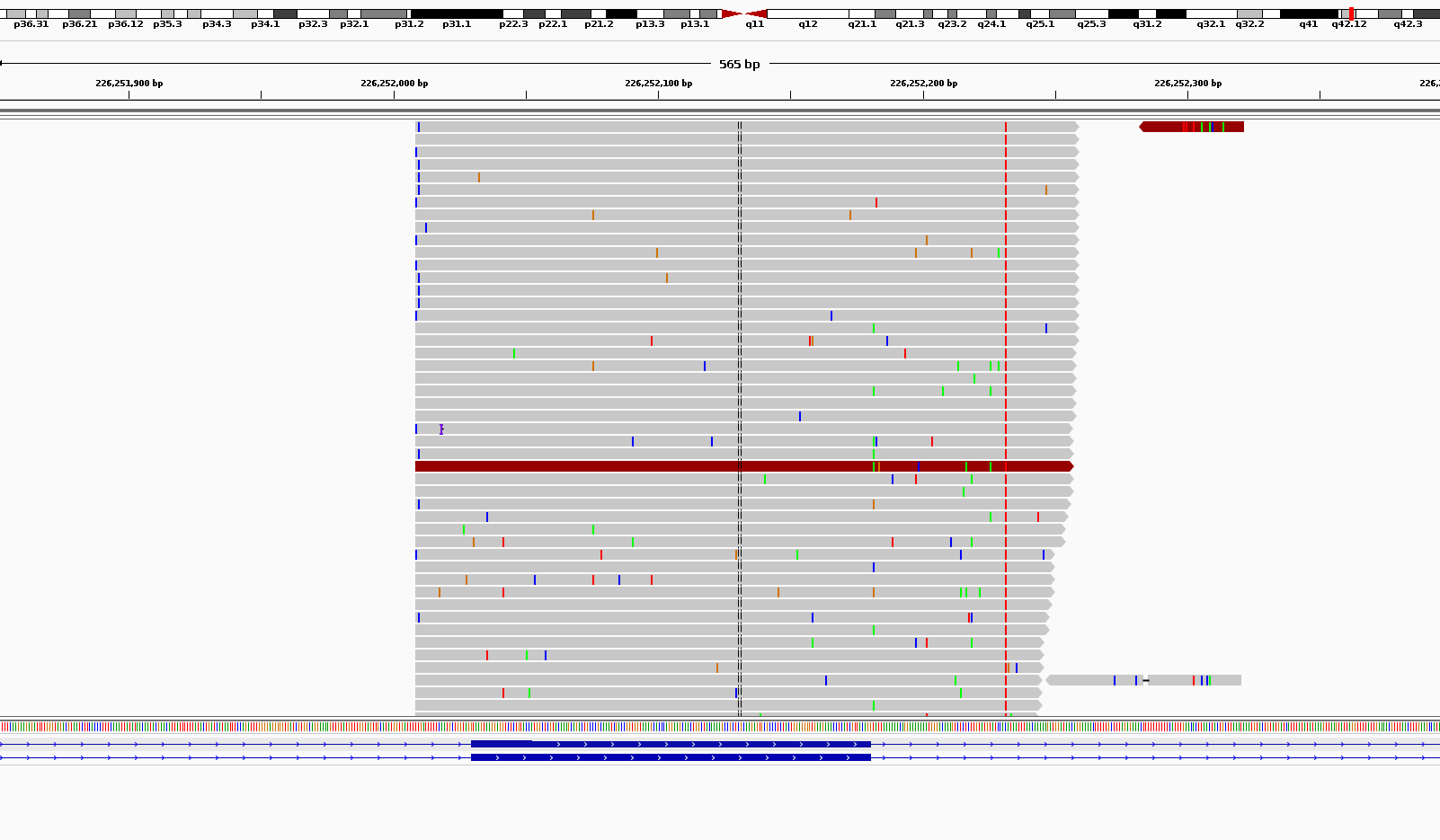
I am trying to hunt for very low frequency substitutions in MiSeq ultra-deep (targeted amplicon) sequencing. The problem is the very vast amount of noises in high coverages. As you can see in the picture below, there are a large number of (partly randomly) scattered pseudo substitutions all around my amplicons. I don't have this problem when I am looking at WES data. I was told that this is somehow normal to see the noise. But the problem is how to distinguish between these noises and real verly low frequency substitutions? Some of them have frequencies near zero and are easy to filter out but what about those with frequencies close to 1%? Also, to get a better estimate of real allele frequencies, I need to consider the amount of noise in calculating the frequencies. For example, if I find a real susbstitution with allele frequency close to 1%, how would I know how much of this 1% is real and how much of it is noise?


First of all make sure you trim your data for quality, especially for MiSeq. There are tools out there, I myself use a script integrated in the PoPoolation toolkit. Second, I would suggest only considering SNPs that are present at least 2-3 times, and discard all singletons.
Thanks Adrian but the question is more about those that already have passed the Q threshold and exist more than a dozen times in coverages around 10,000 (e.g. 24 out of 12,000).
Do you have multiple samples or are these single samples?
Artifacts are likely to be recurrent among multiple samples so if you have multiple samples the best method would be to model the error rates for each SNV at every position in the targeted region and then find SNVs which are outliers of that distribution.
If you have single samples, this problem is more difficult.
There are several samples (more than a hundred actually) with similar phenotypes but from different patients. So it's I would say a combination of both situations.