Entering edit mode

5.9 years ago
williamsbrian5064
▴
510
Hi,
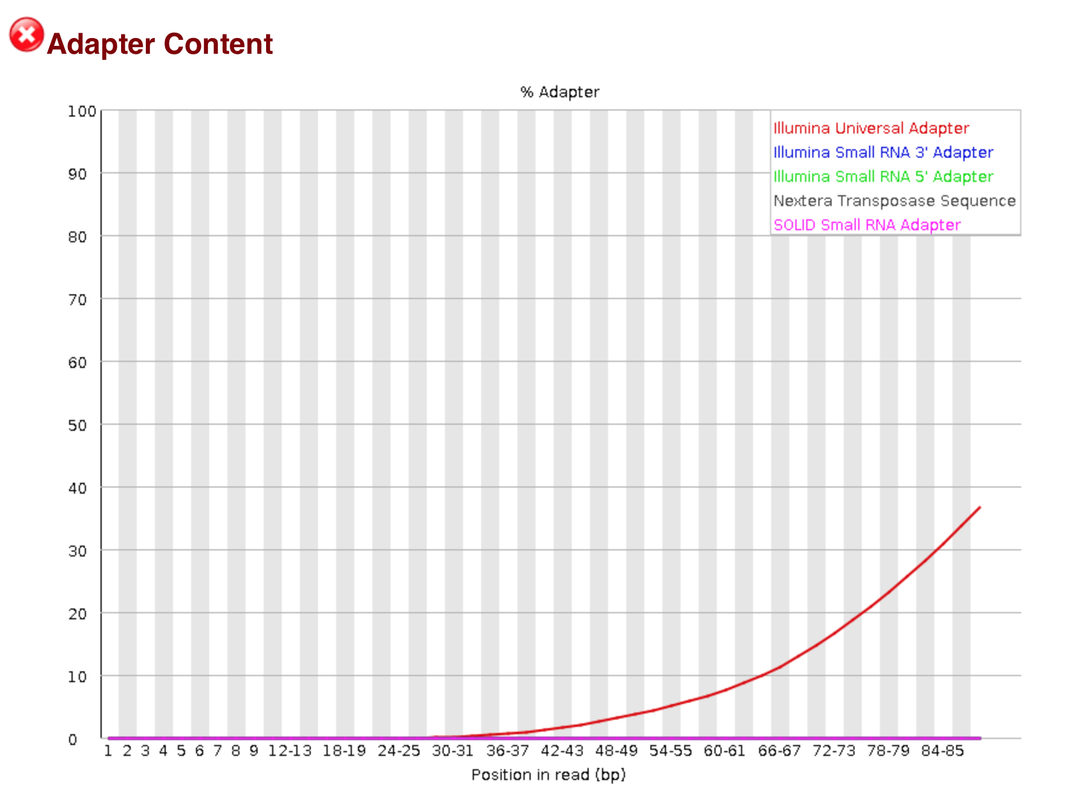
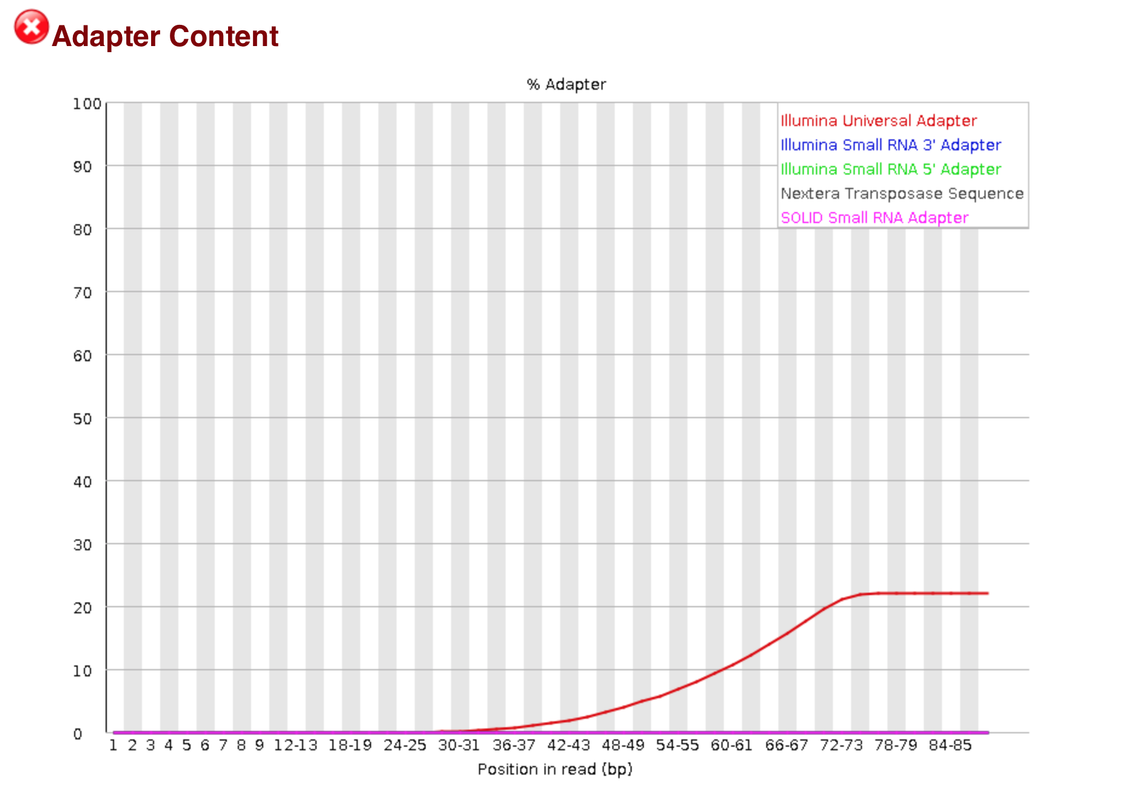
I am attempting to trim some RNA-Seq data. I am having some issues with Illumina universal adapters. I am using trimmomatic to trim the reads and I used trimmomatics TruSeq3-PE.fa file to help trim the adaptors, but there still seems to be a lot of the Illumina universal adapters left. Is this okay? or should I try using a different piece of software. I have used trim galore in the past and that can auto detect adapters. I just don't want to trim away too much data. I know Fastqc doesn't work very well with RNA-seq data
This originally wasn't my project so I am not sure how the library prep was done. I may need to try using trimmomatics TruSeq2-PE.fa, but I am not very confident that will help

I was really referring to some of the flag it will call when looking at RNA seq data. Not saying it can't do RNA-seq data
If this is what you were referring to then no worries. It is a characteristic of RNAseq data.