Entering edit mode

6.2 years ago
Kristin Muench
▴
640
Hello,
I've heard of two different ways of doing batch effect correction:
1. Explicit:
design(myData_collapsed) <- ~ Sex + Genotype
dds <- DESeq(myData_collapsed)
deseqOutput <- results(dds, alpha = 0.05, contrasts = c('Genotype', 'A', 'B'))
2. Implicit:
# make a full model matrix
mod <- model.matrix(~ Genotype, colData(myData_collapsed))
# make a null model to compare it to
mod0 <- model.matrix(~ 1, colData(myData_collapsed))
# calculate SVs
svseq <- svaseq( assay(myData_collapsed), mod, mod0, n.sv = 4)
myData_collapsed$SV1 <- svseq$sv[,1]
myData_collapsed$SV2 <- svseq$sv[,2]
myData_collapsed$SV3 <- svseq$sv[,3]
myData_collapsed$SV4 <- svseq$sv[,4]
# redesign matrix
design(myData_collapsed) <- ~ SV1 + SV2 + SV3 + SV4 + Genotype
# DESeq
dds <- DESeq(myData_collapsed)
deseqOutput <- results(dds, alpha = 0.05, contrasts = c('Genotype', 'A', 'B'))
I only ever see people doing one of the two strategies above. However, it seems to me that it should be possible to do both, just as long as you make sure to include the explicit batches in the design of your model matrix when calculating SVs, like so:
# make a full model matrix
mod <- model.matrix(~ Sex + Genotype, colData(myData_collapsed))
# make a null model to compare it to
mod0 <- model.matrix(~ 1, colData(myData_collapsed))
# make SVs
svseq <- svaseq( assay(myData_collapsed), mod, mod0, n.sv = 4)
# new DESeq matrix design
myData_collapsed$SV1 <- svseq$sv[,1]
myData_collapsed$SV2 <- svseq$sv[,2]
myData_collapsed$SV3 <- svseq$sv[,3]
myData_collapsed$SV4 <- svseq$sv[,4]
design(myData_collapsed) <- ~ SV1 + SV2 + SV3 + SV4 + Sex + Genotype
# perform DESeq
dds <- DESeq(myData_collapsed)
deseqOutput <- results(dds, alpha = 0.05, contrasts = c('Genotype', 'A', 'B'))
Is that correct?
(Or - just realized this - should the null model include the explicit batch effect, e.g. mod0 <- model.matrix(~ Sex, colData(myData_collapsed)) ?)
Thank you for your help! I appreciate all the help I've been getting from BioStars recently, and I hope I can do a good job at documentation for others Googling this issue.

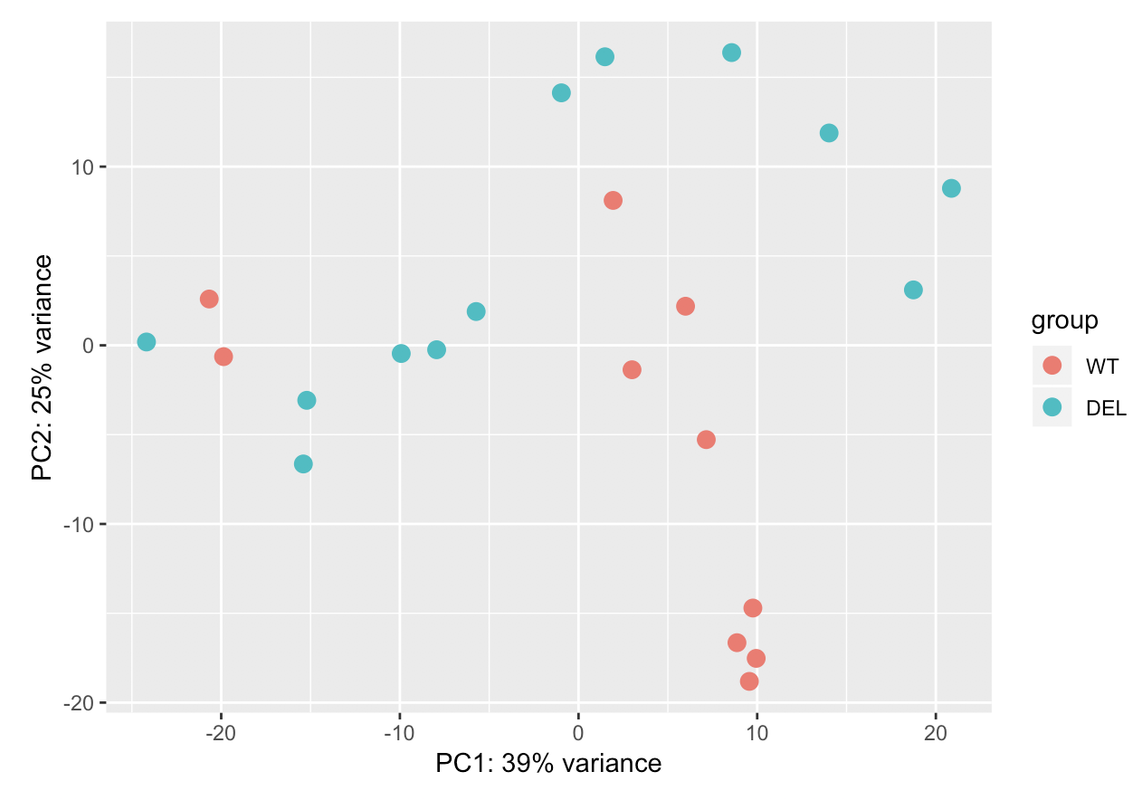
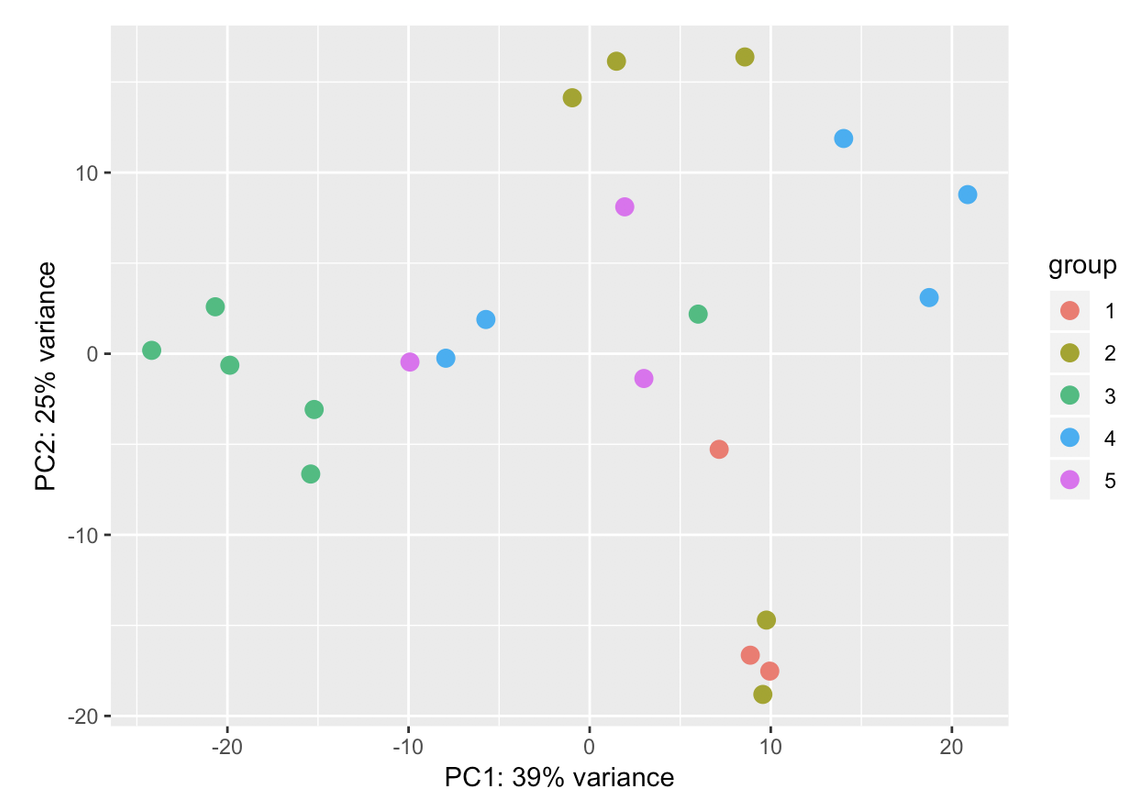
I always go for the explicit when I have the confounding factors measured. Explicit model means you know the batch variables and where they are most likely from and solving the model will tell you how much they are each contributing. In hidden models, you just get a bunch of blobs which you need to dig out what they mean exactly by correlating with known factors.
I also have problems in detecting if the model I have is good as it is
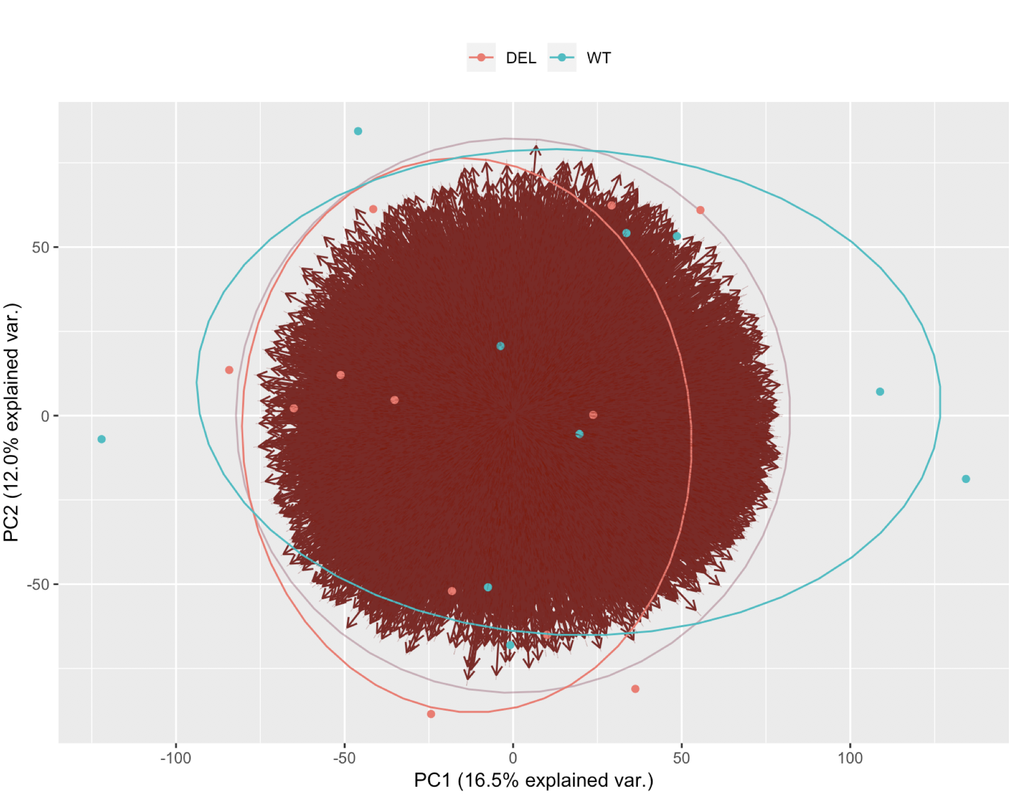
~ sex + conditionor if there is any batch in the data. What it makes me feel a bit confused is the % variance I am obtaining in the PCA plot. Could you help me?Thanks
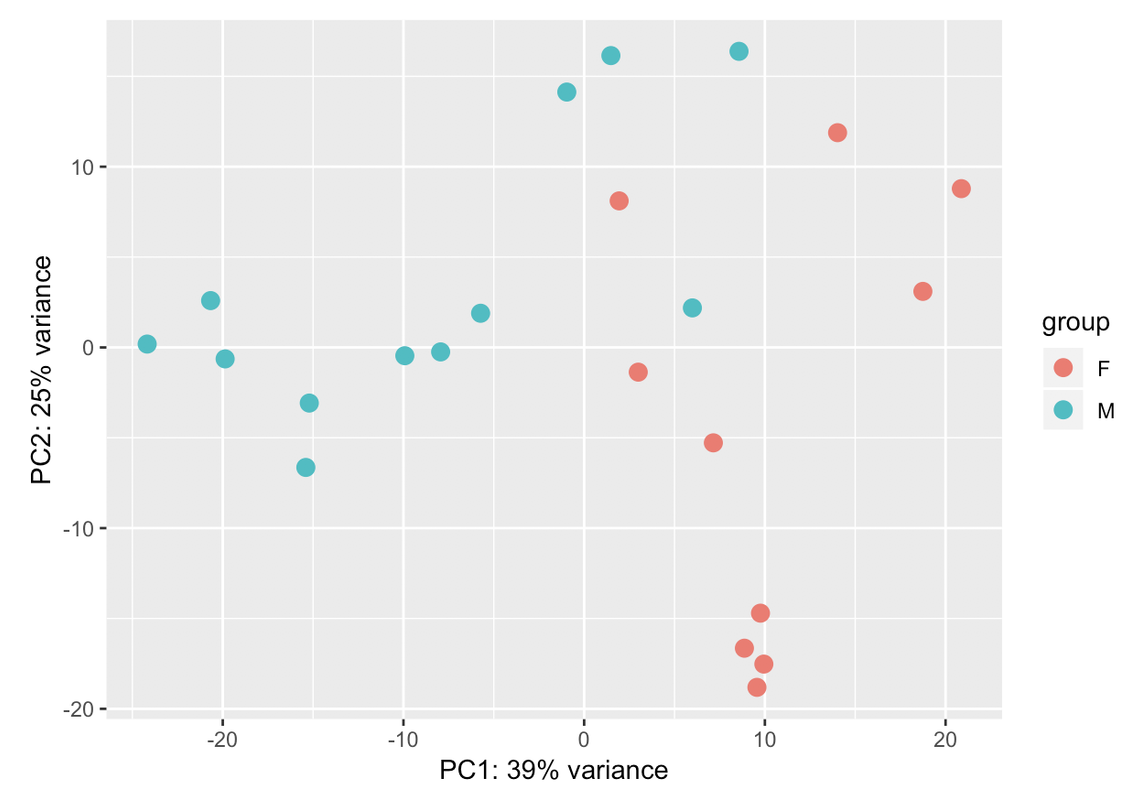
Ciao teresa. I moved your question to a comment. In your PCA bi-plot, the percent variation on PC1 is relatively low (relative to the amount of variation expected with a batch effect, which could be up to 80%); however, the form of the 2 'strata' of samples looks too consistent for it to be based on just natural biological variation. You could try to colour the samples based on other metadata that you have in an attempt to identify a logical reason behind the finding.
Those two groups are separated by gender. When I colour the samples based on it the samples cluster perfectly into both groups: male and female. That's why I have used gender in the design. But I dont know if that is sufficient for the design. I have a question: adding another variable the % assigned would increase? In case that the design is ok with those two variables, would it be a problem presenting that PCA with so low variance in the results?
Thanks a lot Kevin.
This is the resulting plot