Entering edit mode

5.8 years ago
marongiu.luigi
▴
750
Hello,
I have aligned my reads against the reference genome using BWA, I removed the reads with low quality and deduplicated the alignment. How do I now that the results are correct?
Do I need to prove that the reads are mapped correctly by using a second aligner (which should confirm the results of BWA)? This approach I actually did not see it anywhere.
Essentially, I do I know that I am not using garbage? What is the consensus on quality control of the mapping?
Thank you
if you did not mess up BWA parameters nor the pre-processing of reads, I would go by "if it maps, it maps"!
What set you off for this question anyway? Did you notice anything weird in the results (or using the results?)
Well, I wouldn't like that the reviewer comes with some 'below the waterline' question after all this work, so I' rather be sure of the data I gathered. The main problem is that I am aligning against a mix human-virus reference: I need to be sure that the reads are mapped against the virus genome and not against a virus that is actually homologous of a human sequence mistakenly identified as viral. The number of reads mapping the viral genome is very small (and this is comprehensible, assuming that the viruses are very few in the sample), which does not help, and some reads look like split into something else. For instance, this looks OK: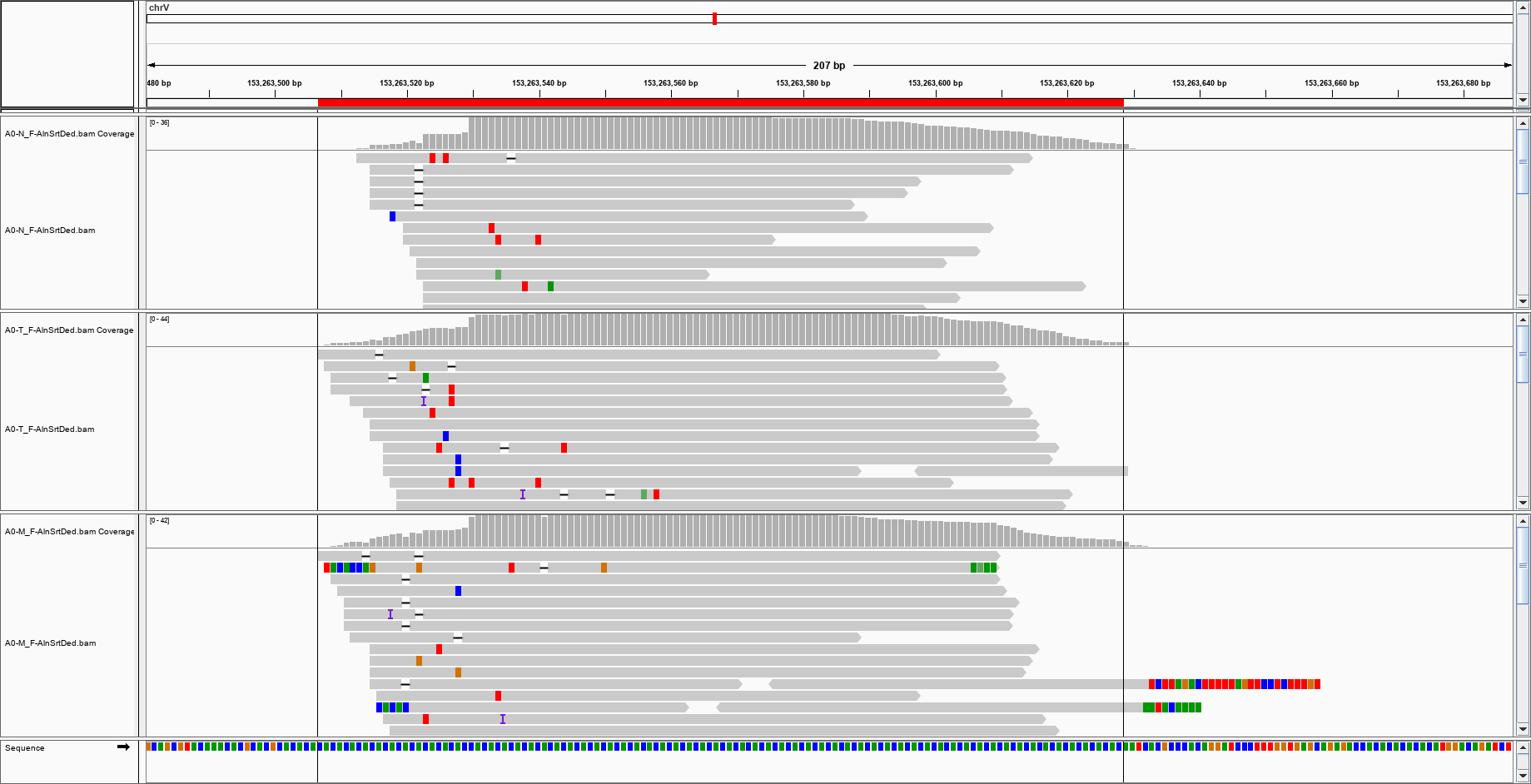 but this one? can I trust it?
but this one? can I trust it?
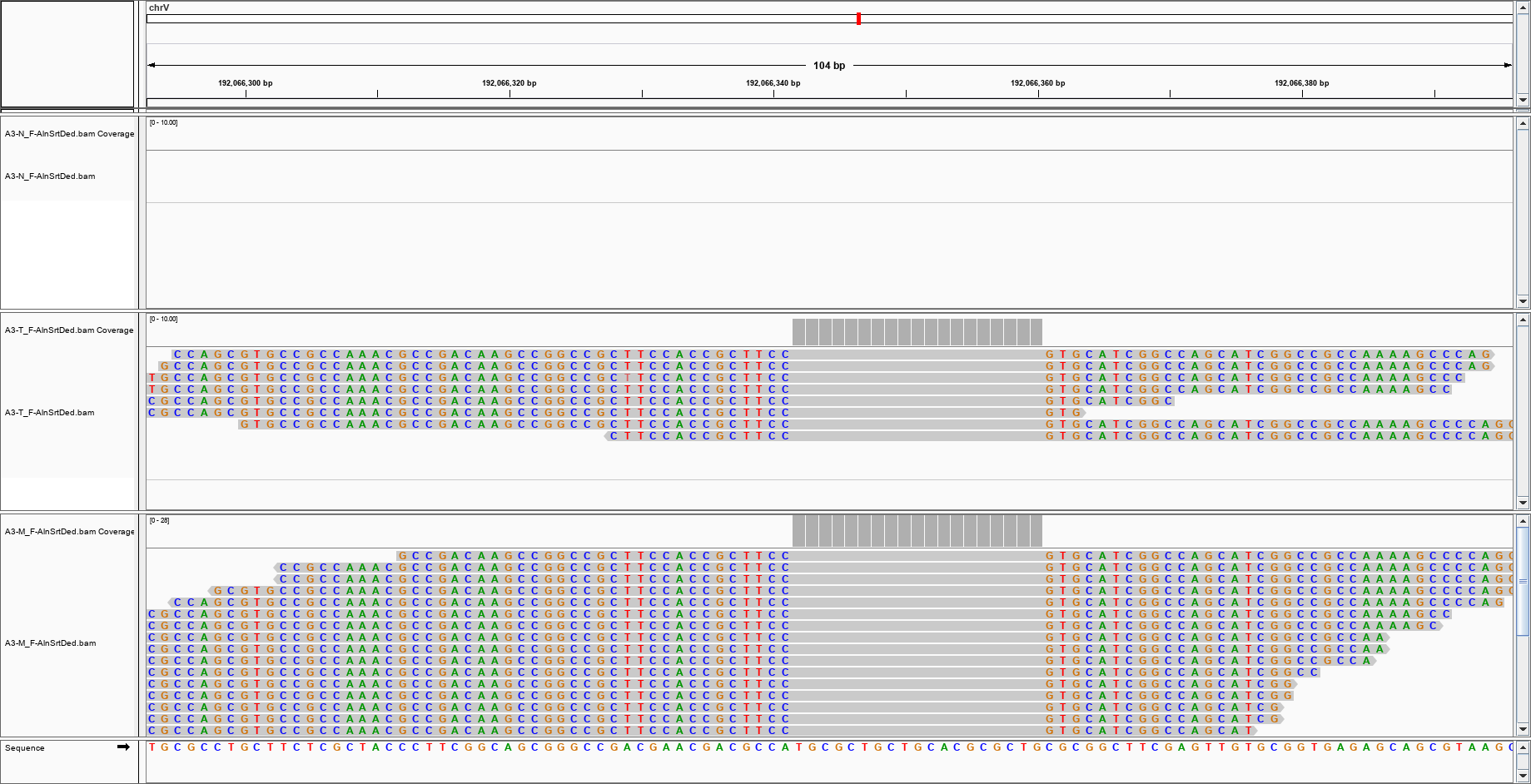 and this?
and this?
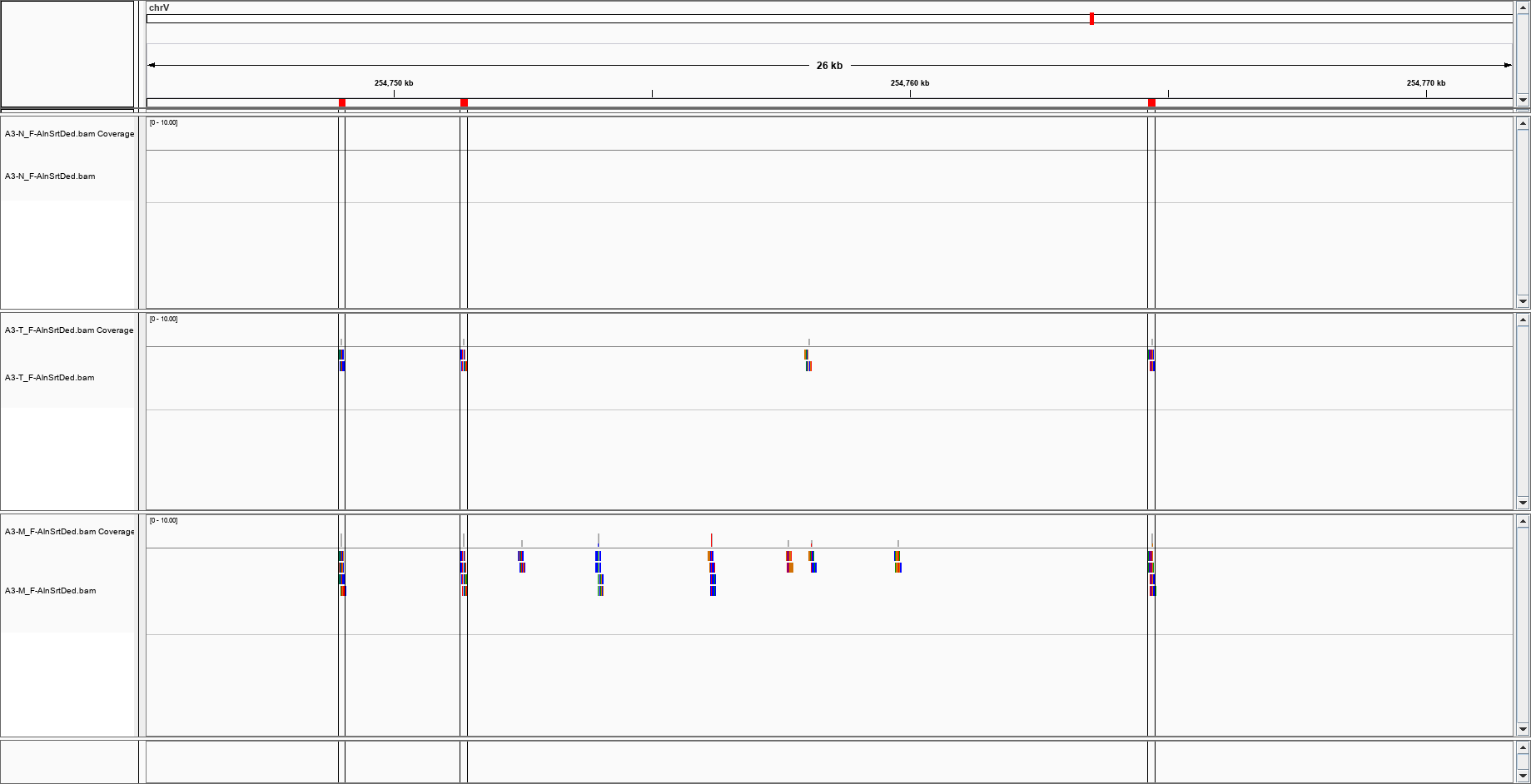 (I have a triad of normal/cancer/metastasis)
(I have a triad of normal/cancer/metastasis)
Are there any regions in the human genome that are very similar to your virus of interest? If you use the viral genome you used as a a reference and cut it up into fake "short read" sequences and align it against the human, where do they go? And what are the alignment scores of the real reads that align to your virus genome compared to the alignment scores of those exact same reads when aligned to the human genome?
If you want to show that the virus genome is a better fit than the human genome, then you gotta show that the alignment scores/edit distances reflect that.
good point! I'll try it...