Entering edit mode

4.0 years ago
marongiu.luigi
▴
710
Hello,
Is there a model (or a reason) why genome with low abundance only show a few portions of the reference genome?
I am doing a WGS on virus sequences; the mean coverage was 25, and for endogenous retroviruses I have such kind of coverage:
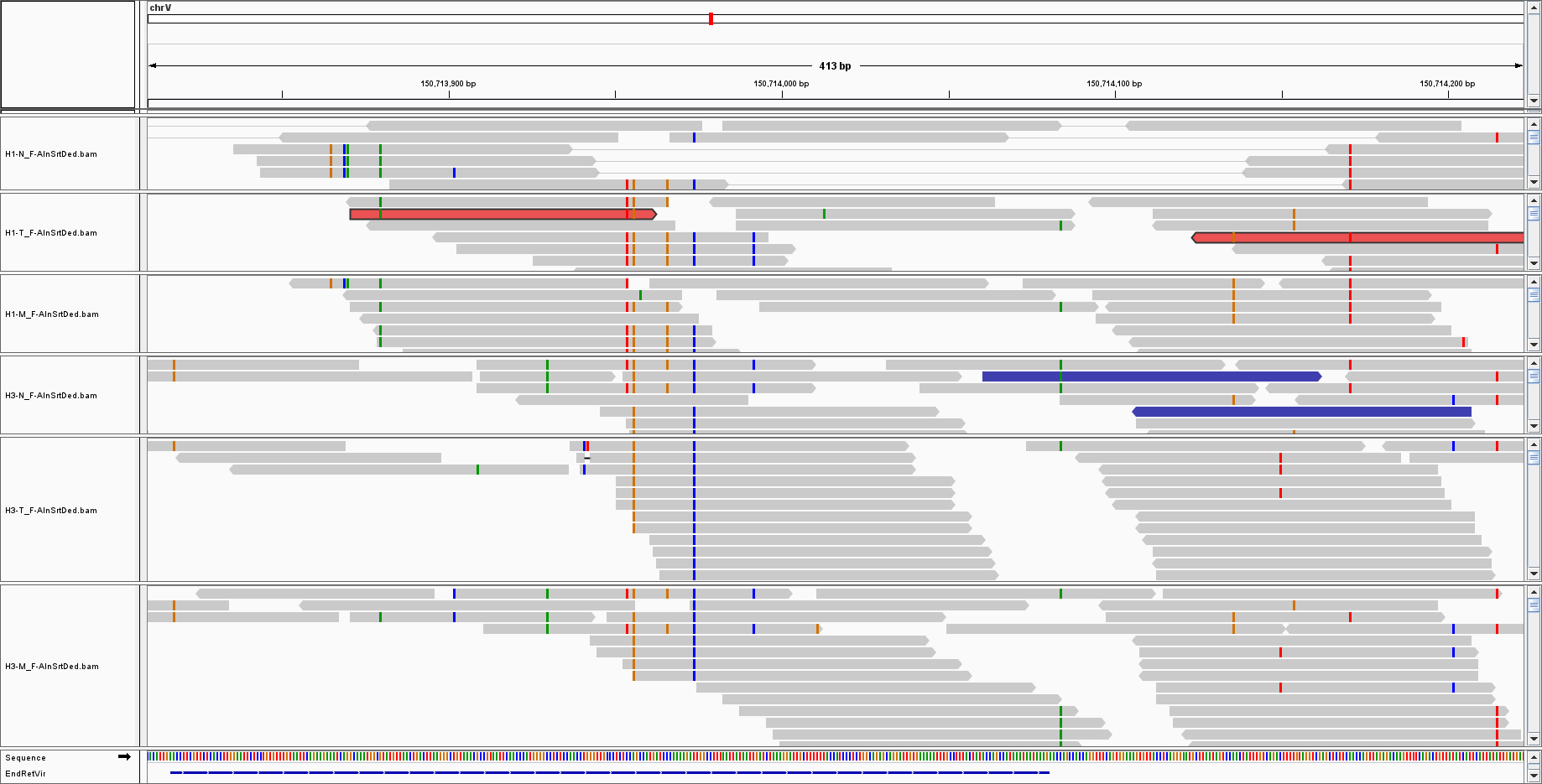
But for other, such as HHV7, I only have sparse reads with low coverage:
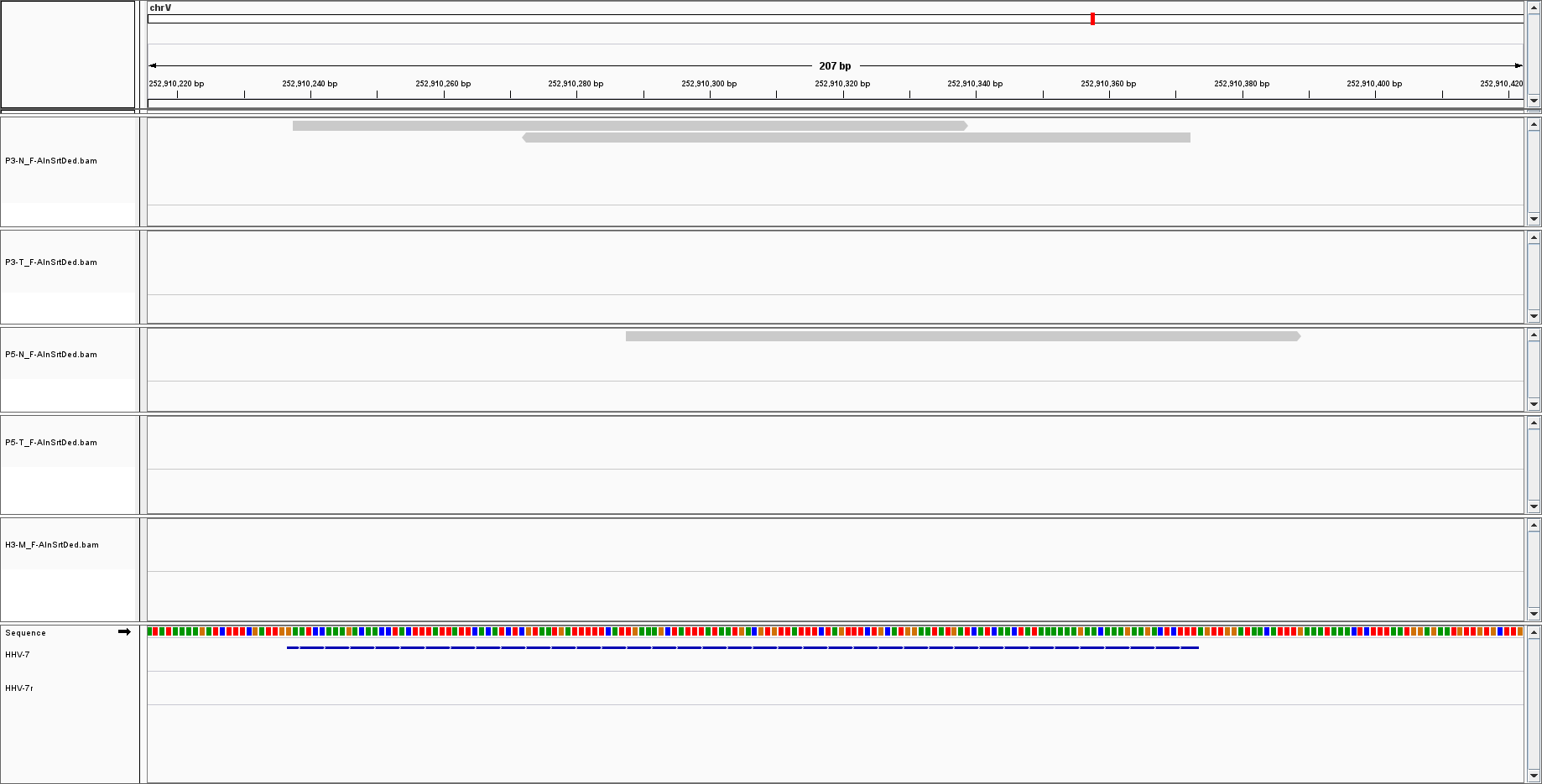
The low coverage I can understand: there is just a little bit of viral DNA, thus there is a low probability of sequencing it.
What I am less comfortable with is the fact the only a portion of the genome is covered.
Is this normal? Is there a model or some paper describing this phenomenon? Are there some factors that increase the sequencing of a certain portion of a low abundance genome rather than others? Thanks
You really have to give more background. What is the setup? A host cell that was infected with a virus? Is it validated that 100% of the cells have the virus integrated? Is it certain that the viral insert is always at the same genomic site? Is the virus only this like 150bp piece?
Sorry: the experiment is a simple WGS of human samples; I am looking at the leftover do not aligned to human chromosomes; the coverage is 25x; sequences are filtered as phred 33 plus blastn and blastx to remove false positive detection of viral sequences. The endogenous retrovirus I am showing is HERV-K113; HHV7 is 145,000 base pairs but i did not say it is integrated. The point is: why did I get 150 bp out of 140 Kb?
Hmm, only reason I could imagine how herpes virus reads get lost is that your reference genome contains the EBV decoy, but this you probably excluded, right? Where did you get the reference genome from?
Er -- no... The decoy is there but I did not include it in the analysis. Perhaps I should have a look at it, thanks for the tip. Anyway, this happens for all other viruses, not only the herpes ones...