Entering edit mode

12.6 years ago
Layla
▴
50
Hi: I have the following table in csv format in Excel:
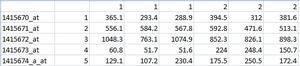
I have been using SAM for getting a list of up and down regulated genes. For that purpose there is an Excel plugin that does the task with the above mentioned file. The problem that I have is that I need to use R for analizing this data, for that there is a package called SAMR. I want to get the list of up and down regulated genes with the names that are in the first column, but no success at all. The manual is available here: http://cran.r-project.org/web/packages/samr/samr.pdf The small program that I made was:
filename<-"test.csv"
y<-c(1,1,1,2,2,2) //I have to do this in this way, but I will extract the
//first row from the csv file later
m<-read.csv(filename,sep=",",row.names=1)
t<-as.matrix(m)
samfit<-SAM(t,y,resp.type="Two class unpaired")
when I put
print(samfit)
I got the following data:
Genes down
Gene ID Gene Name Score(d) Numerator(r) Denominator(s+s0) Fold Change
[1,] g1753 1753 -2.025 -707.725 349.446 0.582
[2,] g1375 1375 -1.038 -272.583 262.7 0.797
[3,] g1302 1302 -0.955 -296.733 310.685 0.739
[4,] g1598 1598 -0.923 -352.725 382.068 0.722
[5,] g1500 1500 -0.913 -442.142 484.177 0.352
but I need the gene names and not that gene ID in the list of genes up and genes down any help? Thanks

It would help to know (1) which organism and (2) what is the source of these gene IDs/names - are they Entrez Gene IDs, for example? Then we can address the topic of ID conversion.